Врожденная эритропоэтическая порфирия (порфирия Гюнтера) — один из наиболее редких типов порфирий, который характеризуется повышенным накоплением предшественников уропорфирина и копропорфирина, преимущественно изомеров I. Это обусловлено тем, что генетический дефект затрагивает синтез уропорфириноген III синтетазы (гидроксиметилбилансинтетазы).
У больных с врожденной эритропоэтической порфирией определяется флюоресценция в ультрафиолетовом свете нормобластов костного мозга и незрелых циркулирующих эритроцитов. Значительное количество порфиринов, выходящих из эритроидных клеток при этом заболевании, объясняет наибольшую степень фотосенсибилизации по сравнению с другими типами порфирий.
Подробное описание врожденной эритропоэтической порфирии сделано в 1911 г. Gunther. Это чрезвычайно редкое заболевание: в настоящее время в мире описано не более 150 случаев. Частота данной нозологической формы не имеет различий в зависимости от пола и расовой принадлежности. Врожденная эритропоэтическая порфирия в подавляющем большинстве случаев диагностируется в детском возрасте.
Молекулярные основы и патогенез врожденной эритропоэтической порфирии
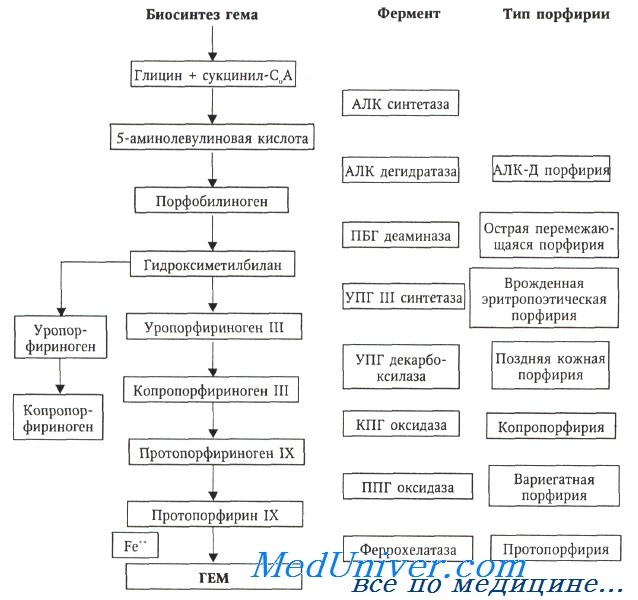
Характер накопления порфиринов при врожденной эритропоэтической порфирии отражает дефект превращения порфобилиногена в уропорфириноген III. Для нормального течения биохимических процессов в данном случае необходимы два фермента: порфобилиногендеаминаза и уропорфириноген III синтетаза.
У больных врожденной эритропоэтической порфирией за счет структурных перестроек гена выявляется снижение активности уропорфириноген III синтетазы и гиперпродукция уропорфириногена I в предшественниках эритроцитов. Активность фермента в эритроцитах и фибробластах снижается до 2-20% по сравнению с нормальными показателями у гомозигот и до 50% — у гетерозигот.
В настоящее время описан ряд различных мутаций гена уропорфириноген III синтетазы, расположенного на 10-й хромосоме.
Несмотря на отчетливое снижение активности уропорфириноген III синтетазы у больных с врожденной эритропоэтической порфирией, синтез гема существенно не страдает. Это объясняется тем, что в норме активность фермента высока, поэтому даже его сниженного уровня достаточно для обеспечения потребности клетки в геме.
Ферментативные нарушения способствуют избыточному накоплению в организме главным образом уропорфирина I (биологически неэффективный изомер, который не может превращаться в гем), в последующем экскретируемого почками. Часто выявляется гемолитический компонент, предположительным механизмом действия которого является фотолиз эритроцитов с избыточным содержанием порфиринов.
Клиническая картина врожденной эритропоэтической порфирии
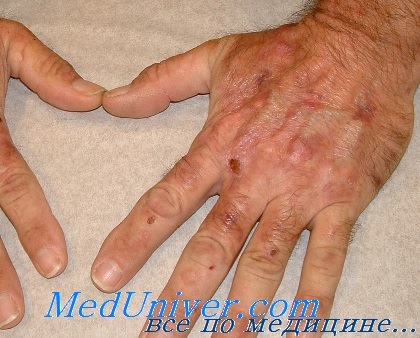
Первым проявлением врожденной эритропоэтической порфирии часто является изменение цвета мочи у детей: от розового до интенсивно красного («цвета бургундского вина»). Наиболее ярко выражена фотосенсибилизация кожи: после загара появляются везикулы или буллы, наполненные флюоресцирующей жидкостью с избыточным уровнем порфиринов. Изменения кожи медленно разрешаются, оставляя после себя пигментные пятна.
Нередко везикулы и буллы нагнаиваются с образованием язв и участков некроза, после которых на коже остаются обезображивающие рубцы. По мере прогрессирования происходит деформация и утрата части органов (пальцев, ногтей, носа, век и ушей). Кожа, на которую не попадали солнечные лучи, остается интактной. Характерен гипертрихоз, сочетающийся с участками отсутствия волос на пораженной коже.
В связи с отложением порфирина в дентине зубы кажутся красными, коричневыми или желтоватыми. В тех случаях, когда изменение цвета зубов незаметно при обычном освещении, определяется красная флюоресценция в ультрафиолетовом свете.
У большинства больных определяются клинические признаки анемического синдрома и часто — спленомегалия.
Данные лабораторных исследований при врожденной эритропоэтической порфирии
В анализе крови выявляется нормоцитарная нормохромная анемия различной степени. Тяжелая анемия встречается редко. Характерны анизоцитоз, пойкилоцитоз, полихромазия, базофильная пунктация, появление ядерных форм эритроцитов. Нередко выявляются признаки гемолиза (ретикулоцитоз, гипербилирубинемия).
Основные изменения в миелограмме выявляются в клетках эритроидного ряда (гиперплазия эритроидного ростка, признаки дизэритропоэза). Продолжительность жизни эритроцитов уменьшена.
Наиболее характерным лабораторным признаком врожденной эритропоэтической порфирии является увеличение мочевой экскреции уропорфирина I. В меньшей степени повышается экскреция уропорфирина III и копропорфиринов I и III. Общая суточная экскреция порфиринов может увеличиваться до 100 мг (в норме — менее 300 мкг/сут).
Профилактика и лечение врожденной эритропоэтической порфирии
Пациенты должны избегать солнечного облучения. На улице необходимо ношение специальной одежды: перчаток, шляп с широкими полями. Обычные солнцезащитные средства неэффективны, так как не защищают от ультрафиолетовых волн длиной около 400 нм, вызывающих порфириновую фотосенсибилизацию.
В большинстве случаев после спленэктомии частично или полностью купируется гемолитический компонент, уменьшаются порфиринурия и фотосенсибилизация. У части пациентов операция недостаточно эффективна.
Применяются также трансфузии эритроцитов, после которых снижается экскреция порфиринов (за счет супрессии собственного кроветворения). Возможно использование режима гипертрансфузий (как при большой талассемии). В таких случаях необходима терапия, предупреждающая развитие гемосидероза (дефероксамин).
Возможно использование длительных курсов лечения активированным углем в больших дозах (по 60 г 3 раза в день). Препарат связывает порфирины, выделяющиеся в желчи, и препятствует их всасыванию в кишечнике. Анемия может уменьшаться при лечении антиоксидантами — альфа-токоферолом и аскорбиновой кислотой.
В тяжелых случаях методом выбора является трансплантация аллогенных стволовых кроветворных клеток. В настоящее время ведется разработка генно-инженерных технологий лечения заболевания.