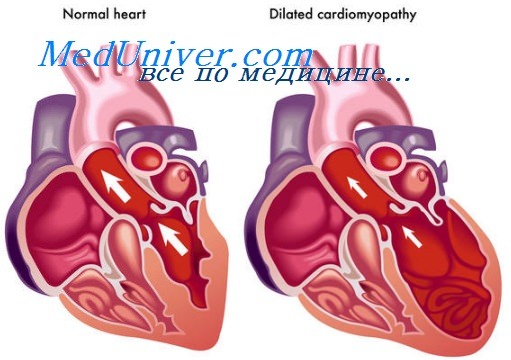
Митохондриальный синдром при ДКМП. Аритмогенная дисплазия правого желудочка
Мутации генов, контролирующих синтез АТФ в процессе окислительного фосфорилирования, могут вызывать полиморфные заболевания, среди которых часто диагностируют ДКМП. Большинство белков, участвующих в окислительном фосфорилировании, кодируются геномом ядра, однако 13 из них — митохондриальными генами. В отличие от ядерных генетических дефектов, мутации митохондриальной ДНК сопряжены с передачей признака по материнской линии. Поскольку митохондриальные хромосомы присутствуют в виде нескольких копий, а мутации зачастую гетероплазматические, поражаются не все копии.
Эти факты в совокупности с данными о различиях в энергетической потребности разных тканей объясняют значительное разнообразие клинических проявлений мутаций митохондриальных генов. Большинство дефектов сопряжено с развитием KMП в сочетании с полиморфными синдромами, такими как Kearns-Sayre, глазная миопатия, MELAS (mitochondrial encephalomyopathy with lactic acidosis, and stroke-like episodes — митохондриальная энцефаломиопатия с лактацидозом и инсульт-подобными эпизодами) и MERRF (myoclonus epilepsy with ragged red fibers — миоклоническая эпилепсия с рваными красными фибриллами), однако некоторые митохондриальные мутации могут привести к преобладанию кардиалыюй миопатии или ее исключительному проявлению.
Установлена ассоциация между гетероплазматическими митохондриальными мутациями и ДКМП. Дефицит энергии может стать тем механизмом, согласно которому мутации митохопдриальных геном и мутации тафаззина вызывают ДКМП.
Аритмогенная дисплазия правого желудочка
Аритмогенная дисплазия правого желудочка — КМП, характеризующаяся прогрессирующей фиброзно-жировой дегенерацией миокарда с прогрессированием его дисфункции, электрической нестабильностью и ВС, наследуется по аутосомно-доминантному типу с неполной пенетрантностью. Более высокая частота заболевания отмечается у мужчин. АДПЖ может развиваться в форме изолированной КМП или в сочетании с двумя связанными заболеваниями, синдромами Naxos или Carvajal, клиническими проявлениями которых являются «рельефная» кожа (ладонно-подошвеппый кератоз), «шерстистые» волосы и нарушения функции преимущественно либо правого желудочка (ПЖ) при синдроме Naxos, либо левого желудочка при синдроме Carvajal. Большая частота распространения АДПЖ в ряде стран, особенно в Италии, не связана с основной мутацией и свидетельствует, вероятно, о более тщательном наблюдении за больными и/или их лучшей информированности о заболевании.
Синдромы Naxos и Carvajal обусловлены рецессивными мутациями плакоглобина, кодируемого геном JUP. Заболевания сердца у больных с этими синдромами сравнимы с таковыми у больных АДПЖ. Причиной изолированной АДПЖ являются доминантно наследуемые мутации пяти генов; гены, расположенные в других локусах и идентифицированные с помощью анализа сцепленных генов, пока неизвестны. Мутации затрагивают десмоплакин (DSP), плакофилин-2 (РКР2), десмоглеин-2 {DSG2) и лесмоколлин-2 (DSC2), каждый из которых кодирует белковый компонент десмосом, высокоорганизованных мембранных структур клетки, которые обеспечивают поддержание структурных и функциональных контактов между соседними клетками. Реже мутации происходят в сердечном рианодиновом рецепторе (RyR2). Связаны ли отдельные мутации в этих генах с клиническими симптомами заболевания, остается неясным.
Для объяснения того, каким образом мутации белка десмосом вызывают АДПЖ, были предложены дна варианта модели. Согласно одному из них, мутации могут стать причиной неадекватной чувствительности десмосом к механическим воздействиям. Другой вариант базируется на анализе процессов передачи сигнала с участием белков десмосом. Обнаружено, что в клетках, экспрессирующих сниженный уровень десмоплакина, повышена локализация плакоглобина в ядре, что мешает канонической Wnt/бета-катенин передаче сигнала и повышает экспрессию генетической программы адипоцитов и фибробластов. Эти результаты позволяют считать, что четкая гистопатология АДПЖ может отражать ренрограммирование клеточной биологии миоцитов при мутациях, вызывающих АДПЖ.