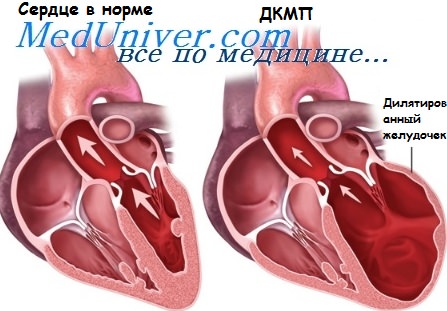
Дилатация сердца. Дилатационная кардиомиопатия без нарушений проводимости
Распространенность идиопатической дилатационной кардиомиопатии в 2 раза превышает таковую при гипертрофической форме, составляя от 2 до 8 случаев на 100 тыс. чел. Приблизительно 50% случаев ДКМП с неясной причиной относят к идионатической форме заболевания. Клиническое обследование родственников первой линии больных идиопатической ДКМП в 30-50% случаев позволяет предполагать участие наследственного компонента.
В большинстве семей (85%) заболевание наследуется как аутосомно-доминантный признак, а в остальных обнаруживают рецессивное наследование признака, сцепленного с Х-хромосомой.
Мутации, вызывающие ДКМП у человека, затрагивают белки, вовлеченные в осуществление целого ряда функций миоцита, в т.ч. генерацию усилия и его передачу, обменные процессы, гомеостаз кальция и регуляцию транскрипции. Несмотря на то что многие гены, связанные с этим заболеванием, уже известны, а ряд мутаций картирован в определенных хромосомных участках, каузальный ген остается неизвестным.
Существенное генетическое разнообразие ДКМП ограничивает ценность селективной молекулярно-генетической диагностики. Анализ ряда связанных с ДКМП генов возможен в сертифицированных лабораториях; исследование ДНК наиболее информативно в тех случаях, когда клинические проявления заболевания ассоциируются с участием конкретных генов. Идентификация генетической мутации в семье, где есть больной ДКМП, как и в случае ГКМП, позволяет прогнозировать развитие болезни у родственников с повышенным риском.
На основании клинических проявлений генетических мутаций, связанных с ДКМП, это заболевание делят на две категории: изолированная ДКМП и ДКМП, для которой характерны экстракардиальные признаки. В свою очередь, мутации в генах, ассоциированные с изолированной ДКМП, подразделяют в зависимости от наличия или отсутствия нарушений проводящей системы сердца.
Дилатационная кардиомиопатия без нарушений проводимости
Мутации, затрагивающие белки, участвующие в обмене кальция, саркомерные белки и другие неидептифициро-ванные макромолекулы, вызывают ДКМП, при которой отсутствуют экстракардиальные проявления или нарушения проводящей системы сердца. Наиболее распространены мутации в генах, кодирующих саркомерные белки|; идентифицированы мутации гена актина (АСТС), тяжелой бета-цепи МНС (MYH7), сердечного тропонина I (TNNI3), сердечного тропонина Т (TNNT2), альфа-тропомиозина (TPM1), титина (TTN) и сердечного тропонина С (TNNC1).
Как и в случае ГКМП, мутации саркомерных белков при ДКМП наследуются по аутосомно-доминантному типу. Признаки заболевания появляются раньше, чем при других мутациях, обусловливающих ДКМП; характерно проявление заболевания в детском возрасте и быстрое прогрессирование. В противоположность этому некоторые взрослые, носители выливающих ДКМП мутаций генов саркомерных белков, имеют нормальную функцию сердца, что свидетельствует о неполной пенетрантности гена.
Гистопатологическая картина ДКМП, связанная с мутациями саркомерных белков, существенно отличается от таковой при ГКМП. Основным признаком, позволяющим отличить первичную ДКМП от конечной стадии ГКМП, является наличие дегенерирующих миоцитов и картина усиленного интерстициального фиброза, протекающего без дезорганизации миоцитов.
Ниши генетических мутаций при ДКМП, затрагивающих саркомерные белки, подобны мутациям, присутствующим у больных ГКМП; в большинстве случаев это аминокислотные замены, вызывающие изменение смысла кодона и небольшие делеции. Более того, локализация мутации в той или иной области гена не связана с ее проявлением. Замены конкретных аминокислотных остатков, обусловленные мутациями при ДКМП или ГКМП, четко различаются, по их близость друг к другу в молекуле белка очевидна. Для изучения механизмов, согласно которым мутации саркомерных белков приводят к ДКМП, использовали анализ мутантных белков методами биофизики и животные модели. В соответствии с полученными данными мутации генов, колирующих саркомерные белки, вызывают ДКМП, влияя на генерацию усилия или его передачу.