Синдром Brugada. Генетические основы синдрома Бругада
В 1992 г. Brugada P. и Brugada R. описали пациентов с подъемом сегмента ST в правых прекорлиальных отведениях (V1-V3) (в отсутствие острого коронарного синдрома), с блокадой правой ножки пучка Гиса, со склонностью к желудочковым тахиаритмиям при структурно нормальном сердце. Сообщалось о семейном наследовании по аутосомно-доминантному типу, предполагая генетическое происхождение болезни. Эта болезнь в настоящее время носит название «синдром Brugada». Частота ее распространения неизвестна, но болезнь чаще встречается в дальневосточных странах.
Типичная аритмия при синдроме Brugada — быстрая полиморфная ЖТ, которая часто переходит в ФЖ; задокументирован также переход в фибрилляцию предсердий. Несмотря на то что синдром Brugada — генетически обусловленное заболевание, клинические проявления (обморок или остановка сердца) в детском возрасте редки и появляются в третьей и четвертой декадах жизни в соотношениях 8 : 1 (мужчины/женщины). Причины таких возрастных и тендерных различий неизвестны. Сердечные события развиваются во время сна или в покое.
Лихорадка, трициклические антидепрессанты и употребление кокаина могут быть пусковыми механизмами сердечных событий у некоторых пациентов.
Диагноз синдрома Brugada затруднен из-за преходящих изменений па электрокардиограмме. Скрытые формы могут быть обнаружены во время пробы с антиаритмиками класса 1С: аймалином (1 мг/кг), флекаинидом (2 мг/кг) или прокаинамидом (15 мг/кг). Автономная модуляция нервной системы может изменить проявления синдрома: внутривенное введение изопротеренола уменьшает ЭКГ-признаки синдрома Brugada, а анетилхолина увеличивает.
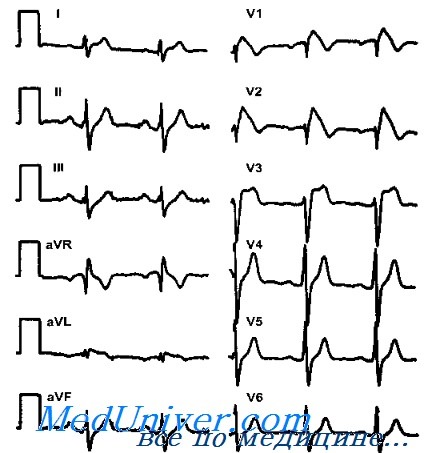
Эта особенность согласуется с тем, что сердечные события при синдроме Brugada наступают главным образом и покое или во время сна. Диагностические критерии синдрома Brugada уточнялись в течение нескольких лет. Выли установлены три типа ЭКГ, однако диагностическим критерием считается только «выпуклая» элевация сегмента ST (тип I). Опасные для жизни сердечные события могут развиться у пациентов с ЭКГ типа «спинка седла» (тип II) с генетически подтвержденным диагнозом.
Генетические основы синдрома Бругада
Первый ген был идентифицирован в 1998 г. как сердечный теп ионного канала натрия (SCN5A). Этот же теп ответственен за генетический вариант LQT3. Хотя синдром Brugada и LQT3 вызваны мутациями в одном и том же гене, существует противоположный эффект этих мутации на ионный ноток натрия: «потеря функции» при синдроме Brugada и усиление функции при LQT3.
Интересно, что о пересечении фенотипов между LQT3 и синдромом Brugada сообщили независимые исследователи, которые подчеркнули функциональную сложность сердечной «болезни ионного канала». Недавно была обнаружена связь мутации SCN5A с ДКМП, что предполагает возникновение структурных нарушений сердца как прямого следствия генетически измененных электрофизиологических свойств кардиомиоцитов. Особенно это относится к пациентам с синдромом Brugada, поскольку наличие структурных нарушений является частью фенотипа.
Было высказано предположение, что вирусная инфекция может служить частью пускового механизма, который сопровождает развитие структурных нарушении у пациентов с синдромом Brugada с мутацией SCN5A или без нее.
В связи с этим при клиническом ведении пациентов необходимо осторожно оценивать ЭКГ у всех пациентов с синдромом Brugada.
Недавно о втором генетическом варианте синдрома Brugada сообщили на примере одной большой семьи с мутацией гена GPD1-L, кодирующего белок, похожий на глицерол-3-фосфат-дсгидрогеназу-1. Функция этого белка в сердце полностью не изучена, по предварительные экспериментальные данные показывают, что мутация гена GPD1-L. уменьшает экспрессию ионного потока натрия.
К сожалению, мутация SCN5A только на 20-25% обусловливает синдром Brugada, подтвержденный клинически, а мутация гена GPD1-L — около 1% случаев синдрома Brugada, о чем свидетельствуют преварительные данные (Priori S.G., Napolitano С, личное сообщение).
Несмотря на то что клиническое значение генетического тестирования при синдроме Brugada ограничено, в случаях, когда оно доступно, эта информация полезна для идентификации «немых носителей» и для доклинической диагностики у членов семьи пробанда.
Учебное видео ЭКГ при синдроме Бругада
При проблемах с просмотром скачайте видео со страницы Здесь