Кисте-стопно-генитальный синдром — аутосомно-доминантное заболевание, характеризующееся укорочением I пальцев кистей и стоп, клинодактилией V пальцев, незавершенным слиянием мюллеровых протоков (при этом возможно удвоение шейки матки и образование продольной влагалищной перегородки) и почечными аномалиями. У мужчин возможна гипоспадия. Кисте-стопно-генитальный синдром обусловлен мутациями гена НОХА13 и наследуется по аутосомно-доминантному типу. Его может вызывать любая мутация, приводящая к гаплонедостаточности гена НОХА13.
НОХА13 — представитель группы факторов транскрипции НОХ, экспрессируемый в дистальных отделах конечностей и мюллеровых протоках. Его роль в слиянии мюллеровых протоков и их дифференцировке до конца не ясна. Синдром Фринса — симптомокомплекс множественных аномалий развития, включающий расщепление верхней губы и нёба, дефекты ЦНС, мальформации мочевыводящих путей, сердца и нарушения слияния мюллеровых протоков. Обычно он детален и наследуется по аутосомно-рецессивному типу.
Большинство хорошо известных на сегодняшний день генетических синдромов, связанных с аномалиями репродуктивных путей, обусловлено недостаточностью ключевых ферментов надпочечников или яичек, наследуемой по аутосомно-рецессивному типу, или дефектом АР. Недостаточность надпочечниковых ферментов, например 21-гидроксилазы, приводит к развитию женского псевдогермафродитизма (кариотип XX).
Недостаточность ферментов, участвующих в синтезе тестостерона, или дефицит АР приводят к мужскому псевдогермафродитизму (кариотип XY).
Олигоспермия (концентрация сперматозоидов менее 5 млн/мл) или азооспермия связана с хромосомными аномалиями в 3-13% случаев. Эти аномалии могут вовлекать половые хромосомы, например 47, XXY; 47, XYY; мозаицизм половых хромосом; структурные аномалии Y-хромосомы. К аномалиям аутосом относятся реципрокные транслокации, робертсоновские транслокации, инверсии и другие структурные аномалии. Чаще всего из хромосомных аномалий, связанных с необструктивной азооспермией, встречается синдром Клайнфелтера (47, XXY).
Наиболее частой генной причиной врожденной обструктивной азооспермии бывают мутации гена регулятора трансмембранной проводимости при кистозном фиброзе.
Синдром Клайнфелтера — самая частая причина гипергонадотропного гипогонадизма у мужчин (высокое содержание ФСГ, низкое — тестостерона). Хотя у большинства больных кариотип 47, XXY, для некоторых характерен мозаицизм. При этом синдроме обнаруживают прогрессирующее истощение популяции зародышевых и стероидсинтезирующих клеток и фиброз половых желез.
Клиническая манифестация характерна для пубертатного периода и представлена аномалиями фенотипа — слишком высокий рост, малые размеры гениталий, редкий волосяной покров на туловище. Обычно таким больным проводят андрогенотерапию. Несмотря на то что у большинства больных выявляют азооспермию, иногда в яичках обнаруживают незрелые или даже зрелые сперматозоиды. Ббльшая часть зрелой спермы, получаемой у больных для искусственного оплодотворения, характеризуется нормальным гаплоидным кариотипом, поэтому их потомство тоже имеет нормальный кариотип.
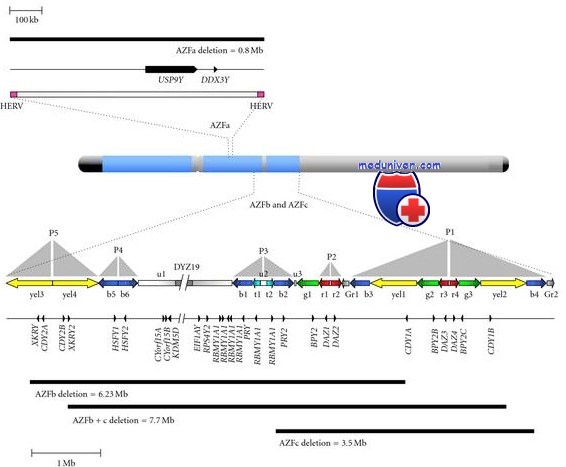
Микроделеции фактора азооспермии при мужском бесплодии. Тестикулодетерминирующий ген SRY локализуется проксимально по отношению к псевдоаутосомному региону короткого плеча Y-хромосомы. На длинном плече Y-хромосомы располагается три региона фактора азооспермии (AZF). В проксимальных отделах длинного плеча, там же, где и ген DAZ, находятся регионы AZFa, AZFb и AZFc. Микроделеции Yq в этих регионах становятся причиной 10-20% случаев азооспермии и 3-10% случаев выраженной олигоспермии.
Делеции AZFa и AZFb, по-видимому, проявляются клинически более тяжело, чем делеции AZFc, которые встречаются чаще. Некоторые мужчины с делениями Yq сохраняют фертильность. Проводят лечение методами искусственного оплодотворения и ИЦИС. Однако потомки мужского пола, рожденные от отцов с микроделециями Yq, тоже, скорее всего, окажутся бесплодными, так как наследуют от них те же микроделеции.