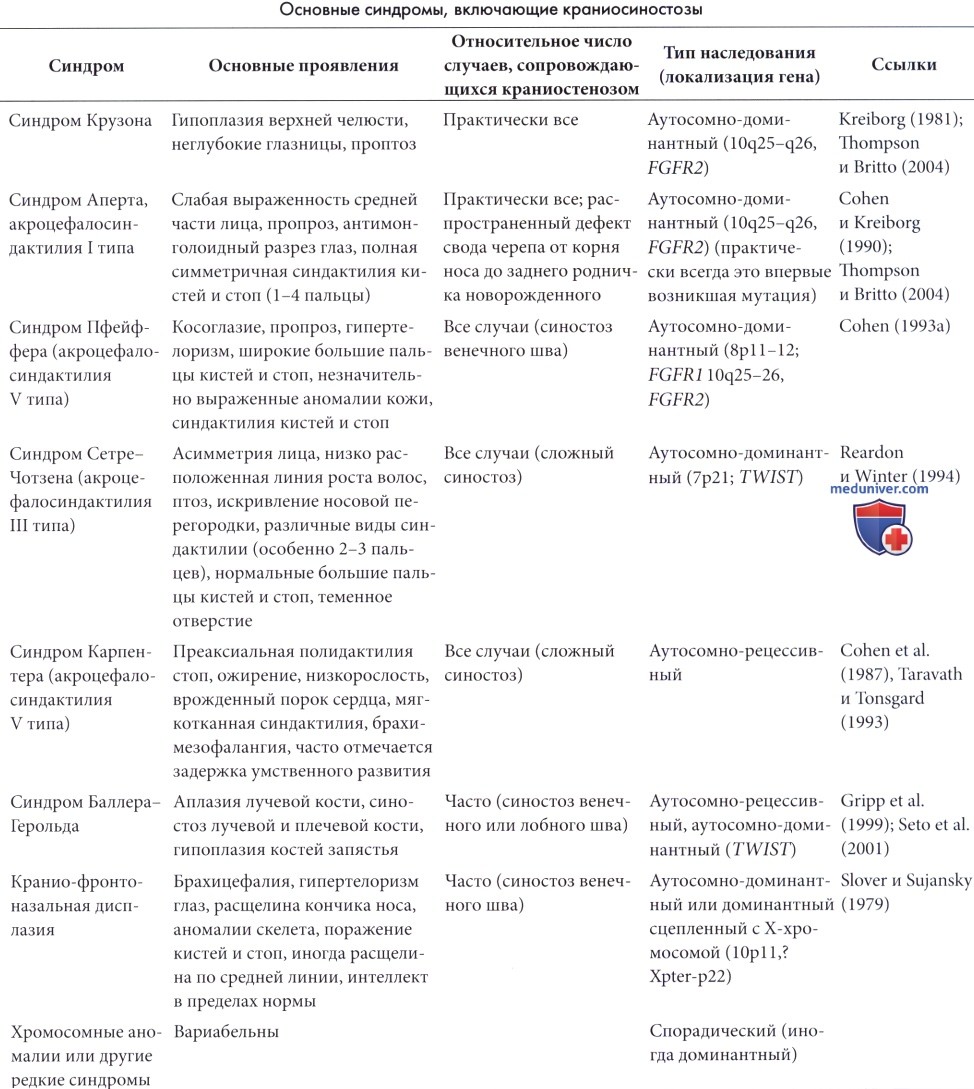
Приблизительно 15-20% краниосиностозов входят в состав различных синдромов, большая часть которых генетически детерминирована. Имеется полный обзор синдромных краниосиностозов (Cohen, 1986, 1988) и подробное описание клинической картины и генетических аспектов (Britto и Reardon, 2004; Thompson и Britto, 2004). Некоторые из наиболее часто встречающихся синдромов представлены в таблице 5.2. Такие синдромы представлены множеством аномалий, одной из которых является краниосиностоз. В рамках одного и того же синдрома тип краниосиностоза может варьировать и не являться определяющим тяжесть заболевания фактором; аномалии органов черепа могут иметь большее значение, так как приводят к нарушениям дыхания, питания и зрения. Диагностика не должна ограничиваться исключительно определением пораженных швов.
На синдромы Крузона и Апера приходится приблизительно по трети случаев синдромных краниосиностозов, а оставшаяся треть встречается при комплексных или редких синдромах, отчасти пока не классифицированных (Le Merrer et al., 1988, Lajeunie et al., 1995).
а) Синдром Крузона. Синдром Крузона характеризуется аутосомно-доминантным типом наследования. Более чем в половине из 61 случая (Kreiborg, 1981) отмечались вновь возникшие мутации; данный синдром объяснял наличие 3% краниосиностозов среди 370 пациентов (Hunter и Rudd, 1977). Установлено, что синдром Крузона и Аперта объясняют 4,8% и 4,5% случаев краниосиностозов при рождении (Cohen и Kreiborg, 1990). Фенотип варьирует даже внутри одной генеалогической линии; часто встречаются слабо выраженные случаи, а внешний вид взрослых может казаться нормальным, но ранние фотографии родителей могут помочь в выявлении минимальных поражений. Ген синдрома Крузона располагается на участке 10q25-q26 (Preston et al., 1994) и кодирует рецептор фактора роста фибробластов-2 (Jabs et al., 1994). Выявлено несколько мутаций данного гена (Reardon et al., 1994; Oldridge et al., 1995).
Существенными проявлениями данного синдрома являются гипоплазия верхней челюсти, неглубокие глазницы и проптоз. Краниосиностоз является постоянным проявлением синдрома, развивается на первом году жизни и чаще всего вначале поражает венечные швы. В итоге происходит поражение всех швов, но форма черепа варьирует от трилистника (Rohatgi, 1991) до скафоцефалии. Часто отмечается выступание черепа в области закрытия переднего родничка. Среди пациентов с синдромом Крузона отмечается выраженная вариабельность проявлений. Часто встречается проводниковая тугоухость поэтому необходимо целенаправленное обследование. Постоянно встречающиеся аномалии внутренних органов и конечностей отсутствуют, но обструкция носовой полости и глотки может приводить к хронической дыхательной недостаточности и формированию легочного сердца (Moore, 1993). Припадки встречаются у 12% пациентов (работы Kreiborg), но задержка умственного развития выявляется редко (3%). Гидроцефалия встречается редко (Marchac и Renier, 1982), но повышение внутричерепного давления было выявлено в 37% случаев, также зарегистрированы случаи хронической грыжи миндалин мозжечка (Cinalli et al., 1995). Аномалии костей запястья и пястных костей встречаются часто и напоминают проявления синдрома Пфейффера.
Учитывая множественные поражения лица, обструкцию дыхательных путей и верхних отделов желудочно-кишечного тракта, хирургическое лечение при раннем проведении дает удовлетворительные результаты.
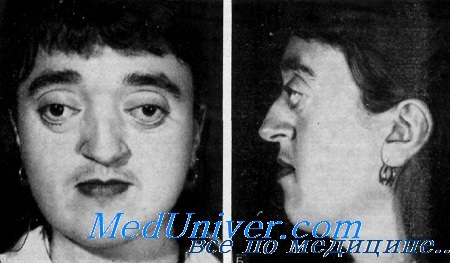
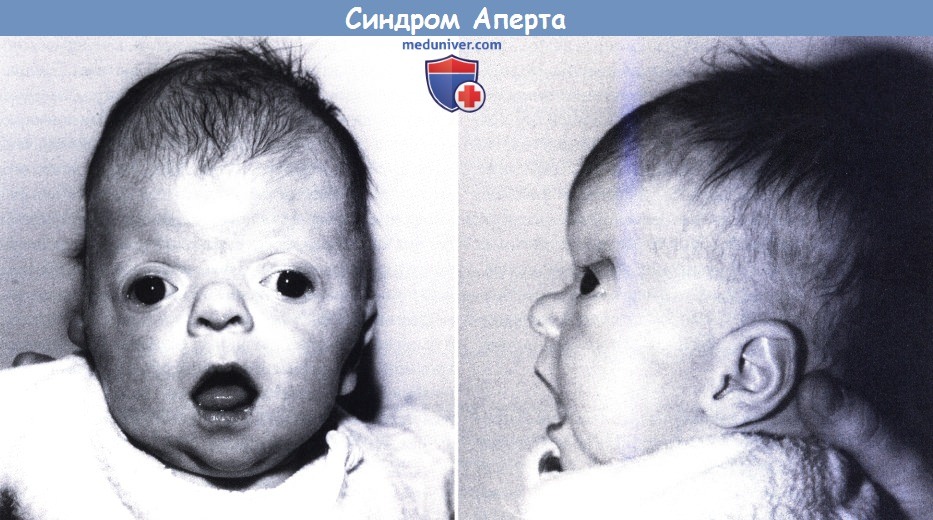
б) Синдром Аперта. Синдром Аперта практически всегда наследуется аутосомно-доминантным путем. Заболевание аллельно синдрому Крузона и вызвано мутацией гена рецептора фактора роста фибробластов-2 (Wilkie et al., 1995). Синдром характеризуется краниосиностозом, мальформациями средней линии лица и симметричной синдактилией кистей и стоп с поражением второго, третьего и четвертого пальцев (Cohen, 1986) (рис. 5.6). Обычно отмечается раннее поражение венечного шва, с формированием высокого брахицефалического черепа, но синостоз имеет вариабельный характер. Распространенный открытый дефект свода черепа начинается от корня носа, продолжается до заднего родничка новорожденных и закрывается до трехлетнего возраста с формированием выпячивания черепа. Передняя черепная ямка короткая, а глазницы неглубокие.
Внутричерепной объем превышает контрольные показатели. Часто встречается расщелина неба. Задержка умственного развития встречается очень часто в очень тяжелой степени. Тем не менее, по результатам одного из исследований задержка умственного развития отмечалась только у половины пациентов (Patton et al., 1988). Достаточно часто встречается гидроцефалия, а аномалии (особенно частичный агенез мозолистого тела) отмечаются у 12% пациентов (Cohen и Kreiborg, 1990). Сращение пятого и шестого шейных позвонков отмечается в 70% случаев; часто встречается глухота и атрофия зрительного нерва. Приблизительно у 10% пациентов выявляются аномалии внутренних органов. Даже при использовании современных реконструктивных методик прогноз неблагоприятный и может быть еще более серьезным при сочетанных аномалиях, таких как аномалии сердца, сколиоз или микроофтальмия.
Синдром Аперта. Гипоплазия верхнечелюстной кости, отсутствие надбровных дуг и экзофтальм.
в) Другие синдромные краниосиностозы. Следующими по распространенности являются синдром Сетре-Чотзена (de Heer et а1., 2005) и синдром Пфайффера (Vanek и Losan, 1982; Zankl et al., 2004). Прогноз для когнитивных функций обычно благоприятный, но синдромы характеризуются заметной вариабельностью неврологических и морфологических проявлений и иногда сопровождаются тяжелыми деформациями.
При синдроме Сетре-Чотзена обычно встречается двусторонний синостоз венечного шва, часто асимметричный (Reardon и Winter, 1994). Аномалии лица, глаз (птоз), внешнего уха и пальцев рук имеют легкую степень выраженности. Синдром имеет преимущественно семейный характер с часто встречающимися малыми формами (Chun et al., 2002).
Синдром Пфайффера также является аутосомным доминантным заболеванием и представлен сочетанием краниосиностоза (преимущественно венечного шва) и характерных аномалий конечностей. Синдром включает широкие и короткие большие пальцы стоп и кистей, симфалангию кистей, частичную мягкотканную синдактилию кистей и стоп. Часто встречается проводниковая тугоухость от легкой до умеренной степени выраженности. Интеллект обычно в пределах нормы. Описано три подтипа заболевания различной степени выраженности (Cohen, 1993b).
Описано большое количество минимально выраженных синдромных краниосиностозов (Thompson и Britto 2004); в настоящее время выделено не менее 67 синдромов (Cohen, 1986), включающих краниосиностозы, и ожидается продолжение. Генетическое консультирование в таких случаях обычно носит неточный характер, но в 7 из 11 случаев недиагностированных синдромных краниосиностозов была выявлена семейная история заболевания (Le Merrer et al.,. 1988), таким образом, вероятность рождения больных детей достаточно высока.
г) Молекулярные аспекты и аспекты развития синдромных краниосиностозов. Последние несколько лет существенное внимание уделяется генетическим аспектам синдромных краниосиностозов (Superti-Furga et al., 2001). Синдромы Аперта, Крузона и Пфейффера связаны с мутациями гена рецептора фактора роста фибробластов-2. Синдром Пфейффера также может быть связан с мутацией гена рецептора фактора роста фибробластов-1. В целом 30 синдромных краниосиностозов расцениваются как моногеные заболевания (Britto и Reardon, 2004). Несиндромные краниосиностозы, особенно венечный синостоз, также могут быть связаны с мутациями гена рецептора фактора роста фибробластов-1 (Lajeunie et al, 1995).
Причины формирования различных фенотипов при мутации в одном и том же гене неясны. Известно большое количество различных мутаций генов рецептора фактора роста фибробластов (Kannu и Aftimos, 2007); существует некоторая корреляция между генотипом и фенотипом, но определенное воздействие дают также эпигенетические факторы и факторы внешней среды. Мутации, являющиеся причиной заболевания, в основном представляют собой миссенс-мутации, а их воздействие основано на «усилении функции»; исключение составляет связанный с синдромом Сетре-Чотзена ген TWIST на 7-й хромосоме, гаплонедостаточность которого служит причинной развития синдрома.
д) Лечение краниосиностозов. В течение последнего десятилетия наметился явный прогресс в лечении краниосионостозов. Многие аспекты лечения невозможно обсудить в рамках данной статьи; для получения исчерпывающей информации следует обратиться к монографии по данной проблеме, вышедшей под редакцией Hayward et al. (2004). В случае сагиттального и одностороннего венечного синостоза и во многих случаях двустороннего венечного и сложного синостоза достигаются удовлетворительные косметические результаты лечения. Наилучшие результаты были достигнуты при раннем оперативном лечении в возрасте до 6 месяцев, но значимая коррекция достигалась позже. Описаны различные комплексные методы лечения (Marchac и Renier, 1982; Marsh и Schwartz, 1983), заключающиеся в обширных хирургических вмешательствах. Общепризнанно, что хирургическое лечение показано в случае повышения внутричерепного давления и/или при начальной стадии атрофии зрительного нерва. В некоторых случаях, таких как сагиттальный синостоз или плагиоцефалия, риск неврологических осложнений невысок, а целью вмешательства является только коррекция формы черепа.
При использовании современных методик лечения изолированных синостозов риск осложнений не столь высок, поэтому хирургическое лечение показано даже при таких относительно доброкачественных формах как сагиттальный синостоз, так как косметические дефекты могут приводить к значимому смущению и затруднению общения в школьном возрасте или позднее. Тем не менее, в ходе недавнего исследования с участием 30 пациентов было продемонстрировано, что у неоперированных пациентов к концу первого десятилетия (9,25 лет) отмечалось нормальное интеллектуальное и психологическое развитие, таким образом, вопрос о необходимости систематического вмешательства остается нерешенным (Boltshauser et al., 2004). Цель лечения должна быть разъяснена родителям. Синдромные синостозы могут быть причиной серьезных отклонений в связи с поражением органов черепа и множеством дефектов скелета и нервной системы. Проблема решается лишь при участии специалистов, знакомых с такими специфическими задачами. Особое внимание уделяется слуху, речи и языку, питанию и психологическим проблемам (Hayward et al., 2004). Необходимо осознать, что лечение не ограничивается хирургическими методами. Важными являются офтальмологические аспекты.
Частота атрофии зрительного нерва снизилась в результате раннего и эффективного хирургического лечения; данное осложнение в 35,5% случаев имело двусторонний и в 9,1% случаев — односторонний характер и было вызвано косоглазием, астигматизмом, гиперметропией или анизометропией; необходима коррекция и наблюдение специалиста (Тау et al., 2006). Часто встречающаяся обструкция дыхательных путей требует консультации ЛОР-специалиста.