Деменция при семейной церебральной амилоидной ангиопатии. Семейные деменции ассоциированные с хромосомой 3
Данное заболевание было описано в 70-е годы в больших семьях британского и датского этнического происхожения, в связи с чем в литературе оно иногда обозначается как «семейная деменция британского типа», «семейная датская деменция» или «офтальмо-отоэнцефалическая гередопатия». Тип наследования аутосомно-доминантный. Заболевание манифестирует на пятом десятилетии жизни и проявляется прогрессирующей деменцией, спастичностью, мозжечковой атаксией, катарактой, снижением слуха. При морфологическом исследовании выявляется тяжелая распространенная амилоидная ангиопатия сосудов мозга, периваскулярные амилоидные бляшки (преимущественно в области гиппокампа) и нейрофибриллярные клубки, что сближает данное заболевание с болезнью Альцгеймера.
Ген заболевания расположен на хромосоме 13q 14 и получил обозначение IMB2B, или BRJ (от англ. British) [Vidal R. et al., 1999]. Продукт гена - интегральный мембранный белок с неустановленной функцией. В британской и датской семьях с данной формой деменции у больных выявлены 2 различных мутации в гене BRI (соответственно, точковая мутация в стоп-кодоне и дупликация участка длиной 10 п.о. со сдвигом рамки), каждая из которых ведет к «проскакиванию» нормального сайта для стоп-кодона и патологическому удлинению мутаитного белка [Vidal R. et al., 1999; 2000].
Синтезируемый в результате белок содержит в своей карбоксильной части чужеродную последовательность из 34 аминокислотных остатков, которая в высвобожденном виде представляет собой амилоидогеиный пептид, становящийся основой формирования нерастворимых амилоидных фибрилл.
Принимая во внимание небольшой размер гена BRI (всего 267 кодонов), в подходящих семьях и изолированных случаях с подозрением па данную редкую форму наследственной деменции наиболее удобным методом прямой ДНК-диагностики болезни является тотальное секвенирование кодирующей области гена.
Семейные деменции ассоциированные с хромосомой 3
В 1999 году R. Davis с соавторами описали 2 семьи с повой формой пресепильной деменции, наследовавшейся по аутосомно-доминаптпому типу. Заболевание начиналось па пятом десятилетии жизни и характеризовалось когнитивным снижением в виде нарушений концентрации и внимания, нарушения зрительно-пространственной ориентировки, расстройств памяти (выраженных в меньшей степени, чем при болезни Альцгеймера). По мере прогрессирования болезни развивалась глобальная деменция, у некоторых больных отмечались также эпилептические припадки. На секции выявлялись особые эозинофильные нейрональные включения в клетках глубоких слоев коры больших полушарий и подкорковых ядер (особенно в области черной субстанции).
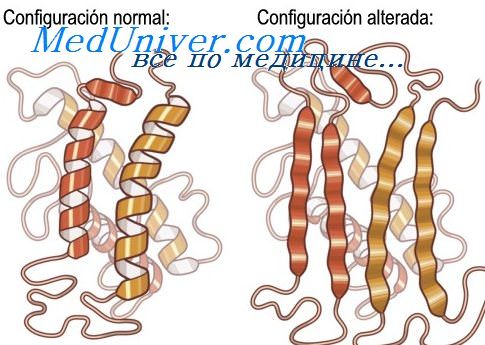
Анализ данных включений показал, что их основным биохимическим субстратом является полимеризованный нейросерпин - 410-аминокислотный нейрональный белок, относящийся к классу ингибиторов сериновых протеиназ (серпинов). В обеих семьях у больных были выявлены точковые миссенс-мутации в гене нейросерпина, расположенном в хромосомном локусе 3q26 [Davis R. et al., 1999]. Показано, что мутантный сейросерпин приобретает нестабильную патологическую копформацию, способствующую его фибриллярной полимеризации и фатальному накоплению в клетке сейросерпин-позитивных нерастворимых комплексов. Таким образом, данный тип наследственной деменции можно отнести к растущему числу «конформационных болезней» (аналогично приоиным болезням, болезни Альцгеймера, болезни Паркинсона, множественным системным атрофиям и ряду других нейрогенеративных заболеваний).
Ведущим проявлением «конформационных болезней» является прогрессирующая дегенерация специфических популяций нейронов, поскольку аккумуляция белковых комплексов несет основную опасность именно для длительно живущих и неделящихся (постмитотических) клеток, наиболее типичным примером которых являются нейроны.