Известна еще одна форма аутосомно-доминантной эпизодической атаксии - так называемая «периодическая вестибуломозжечковая атаксия», характеризующаяся приступами диплопии, осциллопсии, мозжечковой атаксии, тошноты и рвоты. В межприступном периоде отмечается нарушение плавных движений глазных яблок, спонтанный нистагм, угнетение оптокинетического нистагма, а также постепенное появление и нарастание выраженности постоянных коордииаторных нарушений. Данная форма эпизодической атаксии является нозологически самостоятельной, поскольку в соответствующих семьях исключено сцепление с известными локусами эпизодических атаксий 1-го и 2-го типов [Damji К. et al., 1996].
Выбор конкретного гена для исследования определяется особенностями клинической картины, позволяющими достаточно четко дифференцировать между собой основные формы эпизодической атаксии (длительность атактических пароксизмов, особенности их возникновения, реакция на ацетазоламид). Необходимо также принимать во внимание любые пароксизмальные неврологические расстройства у ближайших родственников обследуемого лица (мигрень, эпизоды преходящего гемипареза и т.д.), поскольку полиморфизм клинических проявлений эпизодических атаксий (особенно атаксии 2-го типа) может носить не только межсемейный, но внутрисемейный характер.
В большинстве случаев проводится SSCP-анализ отдельных кодирующих участков гена, после чего наличие мутации в образце с измененными электрофоретическими характеристиками подтверждается прямым секвенированием изучаемого фрагмента ДНК [Denier С. et al., 1999; Jen J. et al, 1999]. Принимая во внимание простое строение гена KCNA1 (один кодирующий экзон, 1448 и.о.), многие исследователи для ДНК-диагностики эпизодической атаксии-1 предпочитают сразу проводить прямое секвенирование кодирующей области гена, амплифицироваиной в трех последовательных реакциях ПЦР [Zuberi S. et al., 1999].
При обследовании больных с предположительным диагнозом эпизодической атаксии-2 на нервом этапе анализа целесообразно исключить экспансию тринуклеотидных CAG-повторов в гене CACNL1A4 в соответствии с протоколом, используемым для прямой ДНК-диагностики СЦА6.
Атаксия-телеангиэктазия
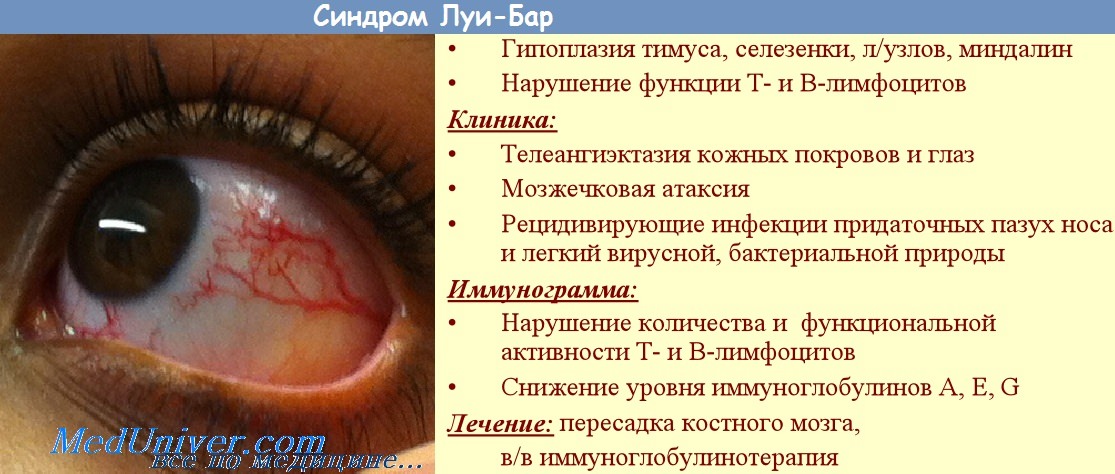
Атаксия-телеангиэктазия (синдром Луи-Бар, синдром Бодера-Седжвика) является относительно частой формой наследственных атаксий, распространенность которой составляет около 1 на 100 000 населения. Тип наследования - аутосомно-рецессивный.
Атаксия-телеангиэктазия характеризуется мультисистемными и весьма полиморфными клиническими проявлениями, которые не ограничены лишь поражением ЦНС, а вовлекают также кожу, сосудистую, иммунную, эндокринную и костную системы [Shiloh Y., 1995; Gatti R., 1998]. Заболевание обычно начинается на 1-2-м году жизни ребенка и проявляется прогрессирущими расстройствами ходьбы и координации движений, к которым могут присоединяться другие неврологическике симптомы (хореоатетоидные гиперкинезы, мышечная гипотония, угнетение сухожильных рефлексов, нарушение психического развития и др.).
Весьма характерными являются изменения мелких сосудов в виде симметричных телеангиэктазий, обычно локализованные на склерах и конъюнктиве глаз, веках, ушных раковинах, спинке носа, области локтевого сгиба и подколенной ямки. К важнейшим диагностическим критериям атаксии-телеангиэктазии относятся частые инфекционные заболевания (обусловленные дисплазией тимуса и дефицитом иммуноглобулинов классов A, G и Е), прогерические изменения кожи и волос, различные онкологические осложнения, скелетные деформации, эндокринные расстройства (гипогонадизм, задержка роста, сахарный диабет).