Определение типа наследования. Группа риска по генетическому заболеванию
После составления родословной и обоснования наследственного характера изучаемого заболевания необходимо установить тип наследования болезни в данной семье. При этом характер сегрегации клинического синдрома сопоставляется с приведенными выше признаками, свойственными всем известным типам наследования. При решении вопроса о типе наследования болезни важно принимать во внимание целый ряд обстоятельств, таких как:
а) возможность неполной пенетрантности мутантного гена («пропуск поколения»);
б) вариабельная экспрессивность мутантного гена (наличие у части родственников «стертых» форм заболевания);
в) близкородственные браки;
г) ранняя смерть одного из родителей (который мог не дожить до возраста манифестации болезни и поэтому характеризовался родственниками как «здоровый»);
д) место рождения родителей и более отдаленных предков (происхождение родителей больного и/или их предков из одного и того же сравнительно изолированного населенного пункта может косвенно свидетельствовать о возможности определенной кровнородственной связи между ними) и т.д.
При наличии нескольких случаев заболевания в семье и несоответствии типа передачи какой-либо определенной модели наследования (менделевской или митохондриальной) может быть сделан вывод о том, что данное заболевание является полигенным (мультифакториальным); это означает, что соответствующие члены семьи наследуют не один основной мутантный ген, а высокую генетическую предрасположенность, которая может привести либо не привести к болези в зависимости от конкретных модифицирующих факторов среды и индивидуальных особенностей генома.
Следует подчеркнуть, что правильно установленный тип наследования уже сам по себе имеет важное диагностическое значение. В качестве примера можно представить типичную ситуацию, при которой в консультируемой семье выявлено несколько случаев прогрессирующей мышечной дистрофии с проксимальным распределением мышечных атрофии и парезов.
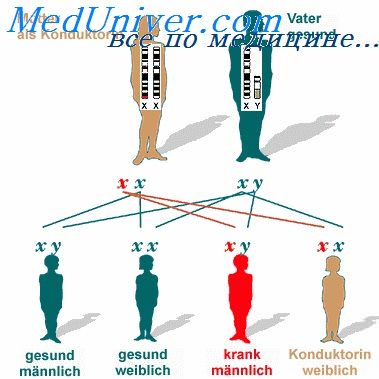
В случае X-сцепленного рецессивного типа наследования такого синдрома следует заподозрить мышечную дистрофию Дюшенна/Бекера, что требует поиска повреждений в гене или белке дистрофине с помощью ДНК-диагностики и иммуногистохимического анализа мышечных биоптатов; с другой стороны, если указанный клинический синдром характеризуется всеми признаками аутосомно-рецессивной передачи, врач должен заподозрить диагноз конечностно-поясной мышечной дистрофии (формы Эрба-Рота) и исследовать другие гены и/или белки мышечного волокна. При этом прогноз заболевания и расчеты генетического риска в консультируемой семье будут различными при разных формах патологии.
Определение круга лиц в изучаемой семье, относящихся к группе риска и нуждающихся в проведении медико-генетического консультирования (т.е. лиц, которые могут являться носителями мутантного гена); расчет конкретной величины генетического риска у консультируемых родственников; осуществление комплекса мер, направленных на профилактику новых случаев заболевания в семье, отягощенной наследственным заболеванием.