Генетика мышечных дистрофий Дюшенна и Беккера. Наследование, молекулярные основы
Подобно муковисцидозу, мышечная дистрофия Дюшенна давно привлекала внимание общества и медиков, поскольку это тяжелое, до настоящего времени неизлечимое, сравнительно частое заболевание с прогрессирующим клиническим течением. Выделение гена, поражаемого при данном Х-сцепленном заболевании, и описание нарушенного белка (названного дистрофином из-за связи с миодистрофией Дюшенна) дали понимание всех аспектов болезни, существенно улучшили генетическое консультирование отягощенных семей и позволили выработать новые подходы к лечению.
Клинический фенотип мышечной дистрофии Дюшенна
Пораженные мальчики здоровы в течение первых нескольких лет жизни, но в возрасте 3-5 лет развивается мышечная слабость, дети начинают испытывать трудности при подъеме по лестнице и при вставании из положения сидя. Ребенок к 12 годам становится инвалидом-колясочником и редко доживает до 20 лет.
Больные умирают от дыхательной или, поскольку также поражается мышца сердца, сердечной недостаточности. На доклиническом и раннем этапах болезни сильно повышается активность креатинфосфокиназы сыворотки крови (в 50-100 раз выше нормы) из-за выхода ее из пораженных мышц. Страдает также и мозг; происходит снижение IQ b среднем на 20 пунктов.
Мышечная дистрофия Беккера
Мышечная дистрофии Беккера также вызвана мутацией в гене дистрофина, но аллели Беккера приводят к значительно более мягкому фенотипу. Считают, что у пациента мышечная дистрофия Беккера, если он сохраняет способность ходить в возрасте 16 лет. Отмечена существенная изменчивость течения болезни, и некоторые пациенты остаются подвижными в течение многих лет. В общих чертах пациенты с миодистрофией Беккера несут мутант-ные аллели, поддерживающие рамку считывания белка, и, таким образом, экспрессируют некоторое количество дистрофина, хотя часто измененного и на малом уровне.
Присутствие дистрофина в мышцах пациентов с миодистрофией Беккера обычно можно обнаружить Вестерн-гибридизацией и иммунофлюоресценцией. В отличие от миодистрофии Беккера, у пациентов с миодистрофией Дюшенна дистрофии не обнаруживается ни одним методом.
Генетика мышечных дистрофий Дюшенна и Беккера
Миодистрофия Дюшенна встречается с частотой около 1 на 3300 живых новорожденных мальчиков, расчетный показатель мутирования 10-4, величина на порядок выше, чем в генах большинства других генетических болезней. Фактически, принимая скорость образования сперматозоидов 8х107 в день, нормальный мужчина производит сперматозоид с новой мутацией в гене миодистрофии Дюшенна каждые 10-11 с!
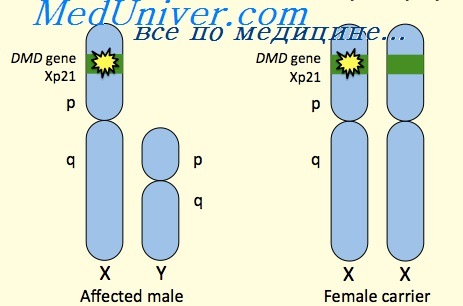
Ранее миодистрофия Дюшенна была представлена как типичное Х-сцепленное рецессивное заболевание, летальное у мужчин, при этом одна треть случаев обусловлена новыми мутациями, а две трети пациентов имеют мать-носительницу мутации. Подавляющее большинство женщин-носительниц не имеют клинических проявлений, хотя примерно у 70% несколько повышен уровень креатинкиназы сыворотки.
В связи со случайной инактивацией Х-хромосомы, тем не менее, у некоторых женщин гетерозигот нормальная Х-хромосома оказывается инактивированной в критической части клеток; около 19% взрослых женщин-носителей имеют некоторую мышечную слабость, а в 8% — жизнеугрожающие кардиомиопатии и серьезные нарушения работоспособности проксимальных мышц. В редких случаях у женщин описана миодистрофия Дюшенна; некоторые имели транслокации X на аутосому, другие только одну Х-хромосому (синдром Тернера) с мутацией миодистрофии Дюшенна в этой хромосоме, и небольшая группа представлена гетерозиготными монозиготными близнецами.
Миодистрофия Беккера составляет приблизительно 15% мутаций в локусе. Важное генетическое различие между этими аллельными фенотипами: если миодистрофия Дюшенна генетически летальна, способность к воспроизводству у мужчин с миодистрофией Беккера достаточно высока (около 70% нормы), чтобы они могли передавать свои гены дочерям. Следовательно, большая часть случаев миодистрофии Беккера унаследована, и только небольшая часть (около 10%) представлена новыми мутациями.
Ген миодистрофии Дюшенна и его продукт
Наиболее примечательная характеристика гена миодистрофии Дюшенна — его размер, оцениваемый в 2300 килобаз, или 1,5% длины Х-хромосомы. Этот огромный ген, подобно гену нейрофиброматоза I типа (NF1) и нескольким другим, находится в группе самых больших известных генов среди всех видов. Высокий показатель частоты мутаций, следовательно, может, по крайней мере частично, объясняться тем, что локус — большая цель для мутаций.
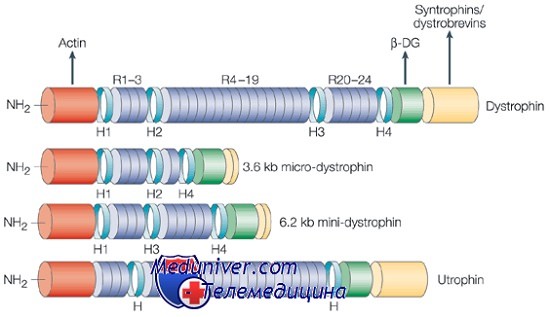
Ген миодистрофии Дюшенна структурно представляет комплекс из 79 экзонов и 7 тканеспецифичных промоторов с различными тканеспецифичными сплайсингами, зависимыми от стадии развития. В мышцах, первичном очаге болезни, большой транскрипт дистрофина (14 килобаз) кодирует огромный белок (427 килодальтон). В соответствии с клиническим фенотипом болезни этот белок в основном представлен в скелетных и сердечной мышце и мозге, хотя большинство тканей экспрессируют по крайней мере одну изоформу дистрофина.
Дистрофии — структурный белок, фиксирующий большой белковый комплекс к мембране клетки. Белковый комплекс дистрофина — настоящее созвездие полипептидов, связанных с генетически различающимися мышечными дистрофиями. Состав комплекса может значительно изменяться в зависимости от изоформ белка как самого дистрофина, так и других присутствующих компонентов, особенно саркогликанов. Комплекс дистрофина выполняет несколько основных функций. Во-первых, его считают существенным для поддержания целостности мышечной мембраны, связывая актиновый цитоскелет с внеклеточной матрицей.
Во-вторых, он играет роль в позиционировании белков в комплексе, обеспечивая их правильное функционирование. Например, комплекс дистрофина необходим в нервномышечных синапсах для соответствующей кластеризации ацетилхолина в ходе развития. Комплекс может также содержать ионные каналы и сигнальные молекулы, что указывает на его возможное участие в опознавании субстратов или других клеток. Хотя функция многих белков в комплексе неизвестна, их ассоциация с болезнями мышц указывает, что они — существенные компоненты комплекса.
Таким образом мутации в некоторых белках комплекса гликопротеидов дистрофина ответственны за аутосомно-рецессивные формы Дюшенно-подобных мышечных дистрофий, пояснично-конечностные и другие мышечные дистрофии.
Посттрансляционная модификация комплекса дистрофина при мышечных дистрофиях Дюшенна и Беккера
Особый интерес представляют пять болезней, вызванных мутациями в гликозилтрансферазах, потеря функции любой из которых приводит к гипогликозилированию а-дистрогликана. То, что для посттрансляционной модификации одного полипептида необходимо пять белков, свидетельствует о значении таких модификаций для нормального функционирования большинства белков вообще и, в частности, о важности гликозилирования а-дистрогликана.
Молекулярный анализ мышечных дистрофий Дюшенна и Беккера
Наиболее частые молекулярные дефекты у пациентов с миодистрофией Дюшенна — делеции (60% аллелей). Распределение делеций в гене не произвольное; они концентрируются в одном из двух регионов в пределах гена, в 5'-половине или в центральном регионе, создающих «горячие точки» делеций. Механизм возникновения делеций в центральном регионе неизвестен, но он включает третичную структуру ДНК и в некоторых случаях рекомбинации между повторяющимися Alu-последовательностями в больших центральных интронах.
Точковые мутации составляют приблизительно треть аллелей и случайно распределены по всему гену.