Этиология и встречаемость синдрома удлиненного QT. Синдромы удлиненного QT (LQT) — разнородная панэтническая группа нарушений, получивших название каналопатий, поскольку они вызываются дефектами в ионных каналах сердца. Распространеность синдромов удлиненного QT приблизительно 1 на 5000-7000 человек. Большинство случаев удлиненного QT вызвано мутациями в пяти известных генах ионных каналов сердца (KCNQ1, KCNH2, SCN5A, KCNE1.KKCNE2).
Генетика, лежащая в основе синдромов удлиненного QT, сложна. Во-первых, существует локусная гетерогенность. Наиболее частый из синдромов удлиненного QT, аутосомно-доминантный синдром Романо-Уорда (MIM №192500), вызван преимущественно мутациями в двух локусах, KCNQ1 и KCNH2, а также содействующим третьим локусом, SCN5A.
Во-вторых, разные мутантные аллели в одном и том же локусе могут вызывать два различающихся синдрома удлиненного QT, синдром Романо-Уорда и аутосомно-рецессивный синдром Джервелла-Ланге-Нильсена (MIM №220400).
Патогенез синдрома удлиненного QT
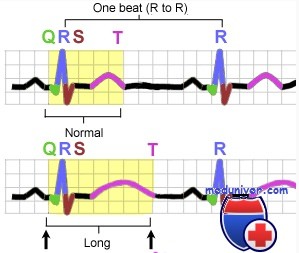
Синдром удлиненного QT вызывается дефектами реполяризации в клетках сердца. Реполяризация — управляемый процесс, требующий баланса между направленным внутрь клетки потоком натрия и кальция и из клетки — калия. Дисбаланс удлиняет или укорачивает длительность потенциала действия, вызывающего соответственно удлинение или сокращение интервала QT на электрокардиограмме.
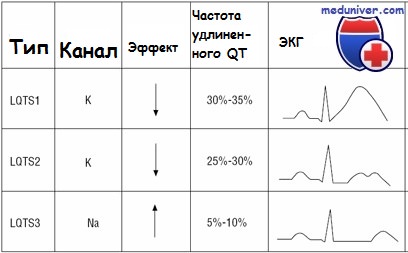
Большинство случаев синдрома удлиненного QT вызваны мутациями с утратой функции в генах, кодирующих субъединицы или полные белки каналов калия (названия этих генов начинаются с KCN). Эти мутации уменьшают реполяризацию, тем самым продлевая потенциал действия клетки и уменьшая порог для последующей деполяризации.
У других пациентов с синдромом удлиненного QT мутации с усилением функции в гене натриевого канала, SCN5A, ведут к повышенному притоку натрия, вызывая аналогичные изменения потенциала действия и эффекты реполяризации.
Фенотип и развитие синдрома удлиненного QT
Синдромы удлиненного QT характеризуются удлинением интервала QT и аномалиями зубца Т на электрокардиограмме, включая тахиаритмию и полиморфную желудочковую тахикардию. Желудочковая тахикардия характеризуется изменением амплитуды и скручиванием комплекса QRS. Полиморфная желудочковая тахикардия связана с удлиненным интервалом QT и обычно заканчивается спонтанно, но может упорствовать и прогрессировать в фибрилляцию желудочков.
При самом частом варианте синдрома удлиненного QT, Романо-Уорда, обмороки из-за аритмии сердца — наиболее частый признак. Если ребенок остается недиагностированным или не получает лечение, синкопальные состояния повторяются и могут быть летальными в 10-15% случаев. Тем не менее от 30 до 50% индивидуумов с синдромом удлиненного QT никогда не имеют синкопальных симптомов. Сердечные эпизоды чаще всего встречаются в возрасте от 9 до 12 лет, уменьшаясь со временем.
Эпизоды могут происходить в любом возрасте, если спровоцированы приемом медикаментов, удлиняющих интервал QT. Нефармакологические триггеры сердечных событий при синдроме Романо-Уорда отличаются в зависимости от ответственного гена. Триггеры LQT1 — обычно адренергические стимулы, включая физическую нагрузку и внезапные эмоции (испуг). Лица с LTQ2 находятся в риске как при нагрузке, так и в покое, а также при слуховых стимулах, например звонок будильника или телефона. Пациенты с LQT3 имеют эпизоды с замедлением сердечных показателей в периоды отдыха и сна.
Кроме того, 40% случаев LQT1 проявляют себя до 10-летнего возраста; симптоматика появляется до 10 лет жизни только в 10% случаев LTQ2 и крайне редко при LQT3. Синдром LQT5 редкий, о течении и триггерах известно меньше.
Синдром удлиненного QT имеет неполную пенетрантность, как с точки зрения электрокардиографических аномалий, так и синкопальных эпизодов. До 30% больных могут иметь интервалы QT, перекрывающиеся с нормальными колебаниями. Варьирующаяся экспрессия заболевания может происходить как внутри семьи, так и между семьями. Из-за неполной пенетрантности для точного диагноза у членов семьи часто необходима нагрузочная электрокардиография.
Синдромы удлиненного QT могут сопровождаться дополнительными данными при медицинском осмотре. Например, синдром Джервелла-Ланге-Нильсена (MIM №220400) характеризуется глубокой врожденной нейросенсорной глухотой в сочетании с синдромом удлиненного QT. Это аутосомно-рецессивное заболевание, вызываемое также определенными мутациями в одном из двух генов (KCNQ1 и KCNE1), участвующих в развитии аутосомно-доминантного синдрома Романо-Уорда.
Гетерозиготные родственники пациентов с синдромом Джервелла-Ланге-Нильсена не глухие, но имеют 25% риск развития синдрома удлиненного QT.
Особенности фенотипических проявлений синдрома удлиненного QT:
• Длинный QTc (>470 мс для мужчин, >480 мс для женщин)
• Тахиаритмия
• Синкопальные эпизоды
• Внезапная смерть
Лечение синдрома удлиненного QT
Лечение синдрома удлиненного QT направлено на предотвращение синкопальных эпизодов и остановки сердца. Оптимальное лечение зависит от идентификации ответственного в данном случае гена. Например, терапия b-адреноблокаторами до начала симптомов — наиболее эффективный метод при LQT1 и, отчасти, при LQT2, но его эффективность при LQT3 незначительна. При лечении b-адреноблокаторами необходимо тщательно проверять соответствие возрастным дозам, не прерывать прием лекарственных средств.
Для больных с брадикардией могут оказаться необходимыми водители ритма; может потребоваться доступ к внешним дефибрилляторам. Имплантируемые кардиовертеры-дефибрилляторы могут быть необходимыми больным с LQT3 или некоторым лицам с синдромом удлиненного QT, для которых проблематична терапия бета-адреноблокаторами, например больным бронхиальной астмой, депрессией или сахарным диабетом, а также пациентам с остановкой сердца в анамнезе.
Некоторые лекарства, например антидепрессивный препарат амитриптилин, фенилэфрин и дифенилгидрамин, или противогрибковые лекарства, включая флуконазол и кетоназол, должны быть исключены из-за их действия, удлиняющего интервал QT или повышения симпатико-тонии. Исключают также виды деятельности и спорта, связанные с интенсивной физической нагрузкой и эмоциональным стрессом.
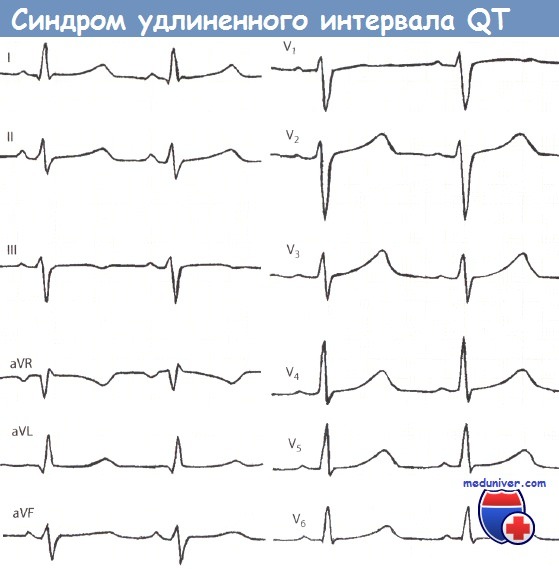
Синдром удлиненного интервала QT (синдром Романо-Уорда).
ЧСС 90 ударов в минуту, длительность QT 0,42 с, относительная длительность интервала QT составляет 128%, откорректированный интервал QTC удлинен и равен 0,49 с.
Риски наследования синдрома удлиненного QT
Лица с синдромом Романо-Уорда имеют 50% шанс родить ребенка с унаследованными мутациями в гене. Поскольку частота новых мутаций низкая, большинство больных имеют пораженного родителя (хотя, возможно, бессимптомного). Чрезвычайно важны и могут оказаться жизнесохраняющими подробный семейный анамнез и тщательная кардиологическая оценка членов семьи. Риск повторения для сибсов пациентов с синдромом Джервелла-Ланге-Нильсена — 25%, как и ожидается при аутосомно-рецессивном заболевании. Пенетрантность изолированного синдрома удлиненного QT без глухоты для гетерозиготных носителей синдрома Джервелла-Ланге-Нильсена — 25%.
Пример синдрома удлиненного QT. А.Б., 30-летняя женщина с синдромом удлиненного QT (LQT), обратилась в генетическую клинику вместе с мужем, поскольку они планируют беременность. Пара хочет знать риск повторения этого заболевания у детей и подходящие методы генетического тестирования и пренатальной диагностики. Женщина также обеспокоена потенциальным влиянием беременности на ее собственное здоровье. Диагноз синдрома LQT установлен в начале третьего десятилетия жизни, когда она проходила обследование после внезапной смерти ее 15-летнего брата. В целом она — здоровый человек с нормальным слухом, отсутствием дисморфических признаков.
У нее никогда не было обморочных состояний. Впоследствии электрокардиографические данные подтвердили диагноз синдрома у нее, ее отца и одной из теток по отцу. Молекулярное тестирование выявило миссенс-мутацию в гене KCNH2, ранее описанную в других семьях с синдромом Романо-Уорда, тип LQT2.
Первоначально пациентка получала бета-адреноблокаторы, но ее кардиологи решили, что низкая эффективность b-адреноблокаторов при LQT2 и летальный случай у ее брата оправдывают имплантацию кардиовертера-дефибриллятора как ей, так и ее пораженным родственникам. Пациентка — первый человек в ее семье, проходящий генетическое консультирование по синдрому удлиненнго QT.