Периодическая болезнь - средиземноморская семейная лихорадка. Диагностика и лечение
Периодическая болезнь, или средиземноморская семейная лихорадка, — наследственное заболевание. Для нее характерны кратковременные острые и самопроизвольно исчезающие подъемы температуры тела, периоды полисерозита, возобновляющегося с нерегулярными интервалами, а также амилоидоз, который в отсутствие лечения приводит к тяжелой почечной недостаточности. Тип наследования — аутосомно-рецессивный. Болезнь встречается главным образом у выходцев из Средиземноморья — евреев-сефардов, турок, apмян и арабов, среди которых частота носительство патологического гена достигает 20 %.
Среди греков, испанцев и итальянцев она несколько ниже. У евреев-ашкенази, немцев и англосаксов периодическая болезнь наблюдается редко, а среди других этнических групп встречается лишь спорадически. В 63-68 % случаев первые клинические проявления возникают до 5-летнего возраста, в 90% случаев — до 20-летнего. Самый ранний возраст начала заболевания — 6 мес. Типичные острые периоды продолжаются 1-4 сут, сопровождаясь лихорадкой и симптомами перитонита (90%), артрит, или артралгии (85%) и плеврита (20%). Перикард и влагалищная оболочка яичка поражаются редко.
К редким проявлениям заболевания относятся также напоминающая рожу эритема кожи, миалгия, спленомегалия, поражение мошонки и ЦНС, геморрагический васкулит и гипотиреоз. У 30-50 % больных в отсутствие лечения развивается АА-амилоидоз почек. Протеинурия за несколько месяцев или лет прогрессирует до нефротического синдрома и почечной недостаточности. Смерть наступает от инфекций, тромбоэмболии или уремии. Амилоидоз наиболее характерен для евреев-сефардов и турок. У армян он развивается реже. Однако среди жителей Армении его частота значительно выше, чем среди армян США. Это указывает на роль факторов окружающей среды в патогенезе амилоидоза.
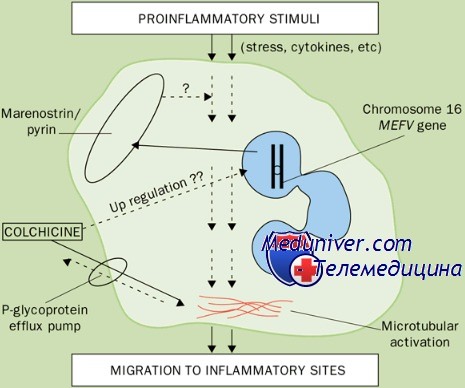
Ген периодической болезни был картирован в небольшом сегменте короткого плеча хромосомы 16 (р13,3) между генами поликистоза почек и синдрома Рубинстайна-Тейби. Затем двум исследовательским группам (международной и французской) удалось выделить и клонировать этот ген, получивший название MEFV. Он принадлежит к семейству генов RoRet, насчитывает примерно 10 тыс. пар оснований и содержит 10 экзонов. Его транскрипт (3,7 тыс. пар нуклеотидов) кодирует состоящий из 761 аминокислот белок пирин (от греч. «огонь»), или маренострин (от лат. «наше море»), который экспрессируется в миелоидных клетках. Обнаружено более 29 мутаций гена MEFV, большинство которых локализуется в последнем экзоне. Все ли они ассоциированы с периодической болезнью, неясно.
Почти у 70 % больных средиземноморского происхождения встречаются те или иные из пяти наиболее частых мутаций (M694V, V726A, M694I, M6801 и E148Q). Анализ гаплотипов и мутаций среди носителей дефектов показывает, что, происходя от общего предка, они в течение столетий разделились. Наиболее частая миссенс-мутация — метионин-694-валин (M694V) — встречается в 30-67% случаев и ассоциируется с тяжелым течением заболевания и высокой частотой амилоидоза. На 2-м месте находится мутация валин-726-аланин (7-35%), ассоциирующаяся с более легким течениям болезни и меньшей частотой амилоидоза. Таким образом, фенотипические различия болезни могут определяться разными мутациями. Клонирование идентификация гена периодической болезни сделали возможной ее диагностику даже там, где она встречается редко и мало знакома врачам. Генетическии скрининг (с помощью ПЦР) теперь проводят в ряде лабораторий.
Однако в таких лабораториях обычно определяют только 5-10 наиболее частых мутаций, пропуская более редкие. Поэтому диагностика периодической болезни все еще основывается на клинической картине, а генетические исследования проводят лишь для подтверждения диагноза. Патогенез обострений болезни остается мало изученным, хотя имеются сообщения о некоторых имунных нарушениях. Особый интерес представляют данные о недостаточности ингибитора (инактивирующего фермента) С5а. Компонент комплемента С5а — анафилатоксин, обладающий высокой хемотаксической активностью. В норме небольшие количества С5а, попадающие в серозные полости, нейтрализуются инактивирующим ферментом еще до того, как вызовут воспаление. Одна из гипотез патогенеза периодической болезни заключается в том, что недостаточность ингибитора С5а (вследствие аномальности пирина) обусловливает накопление его в серозных полостях, что и лежит в основе острых приступов заболевания.
Дальнейшее изучение функции пирина должно пролить свет на его взаимодействие с другими белками, принимающими участие в воспалительной реакции.
Приступы периодической болезни можно предотвратить приемом колхицина в дозе 0,02—0,03 мг/кг (максимум 2 мг) в сутки. Суточную дозу принимают за 1 или 2 раза. Лечение колхицином снижает не только частоту приступов, но и риск амилоидоза, а также уменьшает степень уже имеющегося амилоидоза. Прием колхицина во время беременности, по-видимому, не опасен ни для матери, ни для плода.
В последнее время обнаружено по крайней мере три других синдрома периодической лихорадки, которые следует отличать от периодической болезни. Гипер-IgD периодический лихорадочный синдром (HIDS) — аутосомно-рецессивное заболевание, встречающееся преимущественно в семьях европейского происхождения (голландцев, французов). В его основе лежат мутации гена мевалонаткиназы, локализованного на хромосоме 12 (участок q24). Приступы лихорадки продолжаются более 14 сут и сопровождаются увеличением шейных лимфатических узлов, болью в животе, сыпью, артралгией/артритом и иногда спленомегалией. В крови возрастает уровень белков острой фазы воспаления и IgD (> 100 ЕД/мл).
Специфические методы лечения неизвестны, хотя иногда помогают глюкокортикоиды. Периодический синдром, ассоциированный с рецептором ФНО (TRAPS), ранее носил другие названия: семейная ирландская лихорадка, семейная периодическая лихорадка и аутосомно-доминантная повторяющаяся лихорадка. Описано лишь несколько случаев этого синдрома в семьях ирландского и шотландского происхождения. В основе лежат мутации гена, расположенного на хромосоме 12 (участок р13) и кодирующего рецептор ФНО 1-го типа. У больных возникают кратковременные (4-6 сут) подъемы температуры, сопровождающиеся болью мышц живота. Могут иметь место сыпь, конъюнктивит и односторонний периорбитальный отек.
Иногда возрастает содержание белков острой фазы воспаления, но наиболее специфичный признак — низкий уровень растворимого рецептора ФНО 1-го типа в сыворотке крови. Для лечения применяют глюкокортикоиды. Показана терапевтическая эффективность этанерцепта (рекомбинантного рецептора ФНО). Недавно описано заболевание, названное синдромом периодической лихорадки с афтозным стоматитом, фарингитом и аденитом (PFAPA). У некоторых больных наблюдалась и артралгия. В большинстве случаев симптомы исчезали уже после однократного приема преднизона (1-2 мг/кг). Обычно этот синдром развивается до 5-летнего возраста и через 4-8 лет самопроизвольно исчезает. Неясно, имеет ли он инфекционную природу или связан с иммуногенетическими нарушениями.