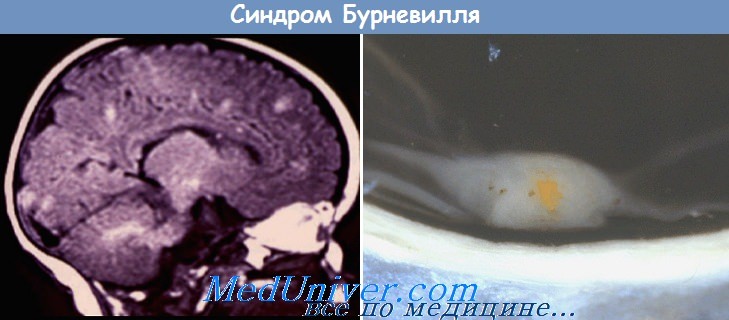
Туберозный склероз (болезнь Бурневиля—Прингля). Заболевание описано D. Burneville в 1862 г. В 1880 г. J. Pringle выделил его в самостоятельную нозологическую форму. Заболевание наследуется по аутосомно-доминантному типу с неполной пенетрантностью мутантного гена. Частота патологии среди умственно отсталых составляет 0,3 %.
При патоморфологическом исследовании в мозге обнаруживают узелки или бугорки белесоватого цвета, склонные к обызвествлению. Бугорки могут локализоваться в коре и белом веществе полушарий, в стволе мозга, мозжечке, спинном мозге. Гистологически в мозговой ткани определяются разрастание гли-альных волокон, а также атипичные гигантские мультиполярные клетки, встречающиеся главным образом в тубероидных очагах. Часто обнаруживаются опухоли внутренних органов.
Основными клиническими проявлениями туберозного склероза являются изменения кожи, судорожные припадки, психические и эндокринные расстройства, пороки развития внутренних органов. Симптомы заболевания чаще появляются на 1-м году жизни. У детей возникают полиморфные судорожные приступы: большие развернутые припадки, малые припадки по типу абсансов, кивков, салаамовых судорог. Больные резко отстают в психическом развитии, так что в более поздней стадии на первый план в клинической картине выступает снижение интеллекта до степени идиотии.
Нередко обнаруживаются гидроцефалия, пирамидные и экстрапирамидные симптомы, адипозо-генитальный синдром. В возрасте 2—6 лет, а изредка и на 1-м году жизни, появляются разнообразные кожные изменения. Наиболее характерны аденомы сальных желез, располагающиеся на щеках в форме «бабочки» и имеющие вид розовато-желтоватых или красных папул. Встречаются также пигментированные и депигментированные пятна, подкожные фибромы, папилломы, пигментированные бляшки. В поясничной области иногда обнаруживается своеобразная шероховатость («шагреневая кожа»).
Среди других симптомов следует отметить опухоли внутренних органов, прежде всего почек и сердца (рабдомиома). На глазном дне определяется новообразование в виде тутовой ягоды, у 50% больных — застойные диски зрительных нервов.
При рентгенологическом исследовании черепа у 50% больных обнаруживаются петрификаты в области боковых и III желудочков, а также в мозжечке. На ЭЭГ — диффузные и очаговые изменения биоэлектрической активности.
Заболевание прогрессирует медленно, но вследствие тяжелого слабоумия больные нуждаются в постоянном наблюдении.
Туберозный склероз следует дифференцировать от других форм олигофрении, эпилепсии и заболеваний, приводящих к органической деменции.
Лечение симптоматическое: противосудорожное, седативное, общеукрепляющее.
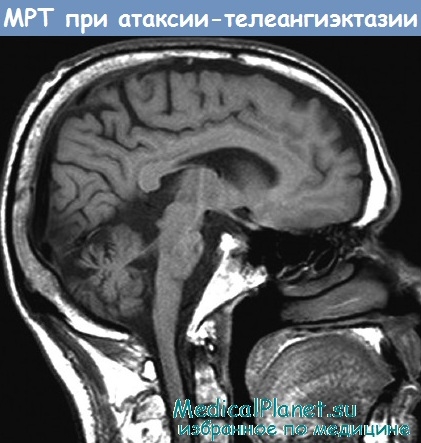
Атаксия-телеангиэктазия (синдром Луи-Бар)
Заболевание описано D. Louis Bar в 1941 г. Наследуется аутосомно-рецессивно. Определенную роль в его патогенезе играет нарушение Т- и В-иммунных систем.
При патоморфологическом исследовании обнаруживают дегенеративные изменения в мозжечке — уменьшение количества и дистрофические изменения клеток Пуркинье, атрофию белого вещества. Отмечается избыточное ветвление мелких вен мягких мозговых оболочек мозжечка, сосудов кожи и слизистых оболочек. Вилочковая железа маленькая, отсутствует деление ее на корковое и мозговое вещество; количество телец Гассаля и лимфоцитов резко снижено. Размеры селезенки и лимфатических желез также уменьшены.
Важным, а иногда и самым ранним, признаком заболевания являются сосудистые изменения на конъюнктиве глазных яблок, слизистой оболочке рта, на коже туловища и конечностей в форме телеангиэктазий — характерных сосудистых паучков. Эти сосудистые образования могут иметь артериальное и венозное происхождение. У многих больных описывают также пигментированные пятна цвета кофе с молоком на коже, участки депигментации, кератоза, склеродермии.
Наряду с кожными и неврологическими проявлениями при синдроме Луи Бар наблюдается склонность к инфекционным заболеваниям легких и дыхательных путей, хроническое течение которых может привести к бронхоэктазиям, пневмосклерозу. Частые заболевания системы органов дыхания обусловлены имеющейся у больных дисгаммаглобулинемией.
Дети отстают в росте и массе. У многих из них имеются диспластические черты развития: незаращение дужек позвонков, фридрейхоподобные стопы и др.
При рентгенологическом исследовании черепа у многих больных обнаруживается усиление пальцевых вдавлений. На ЭЭГ — диффузные изменения биоэлектрической активности. При пневмоэнцефалографии выявляется атрофия мозжечка. В крови отсутствует иммуноглобулин-А и снижено содержание у-глобулина.
Течение заболевания неуклонно прогрессирующее. Летальный исход обычно обусловлен заболеваниями легких и развивающейся легочно-сердечной недостаточностью.
Дифференциальный диагноз следует проводить с врожденными и приобретенными атаксиями, с селективной недостаточностью иммуноглобулина-А.
Для лечения синдрома Луи-Бар помимо симптоматической терапии в последние годы применяется пересадка вилочковой железы, разработанная во 2-м Московском государственном медицинском институте им. Н. И. Пирогова. Тимус с частью грудины экстирпируют у погибшего новорожденного и подсаживают под кожу бедра. В результате операции частично корригируется иммунодефицитное состояние, что приводит к улучшению состояния больных.