Пароксизмальный паралич описан в 1882 г. русским врачом И. Шахновичем. В последующие годы было установлено, что существует несколько форм патологии, неоднородных по генетическому происхождению и клиническим симптомам: гиперкалиемическии пароксизмальныи паралич (болезнь Гамсторп), нормокалиемический пароксизмальный паралич и гипокалиемический паралич (болезнь Вестфаля). У детей раннего возраста встречается только одна форма — периодический нормокалиемический паралич, который наследуется аутосомно-доминантно с полной пенетрантностью патологического гена.
При патоморфологическом исследовании изменений в нервной системе не обнаруживают. При исследовании мышцы, биопсированной во время приступа, выявляется отложение гликогена.
Заболевание проявляется в возрасте 2—5 лет, иногда позднее, пароксизмами, нарастающей слабостью мышц конечностей, туловища, лица, жевательных мышц. В процесс вовлекается и дыхательная мускулатура, что при быстром нарастании мышечной слабости может привести к асфиксии и летальному исходу. Параличи обычно симметричные, характеризуются выраженной мышечной гипотонией, снижением или отсутствием сухожильных и надкостничных рефлексов. Механическая или электрическая возбудимость мышцы также снижается или исчезает (так называемая трупная реакция). Электромиографически во время приступа определяются короткий полифазный потенциал, снижение амплитуды кривой. Проксимальные мышцы поражаются в большей степени, чем дистальные. Чувствительных расстройств не наблюдается. Во время приступа отмечаются усиленное потоотделение, гиперемия лица, слюнотечение, мидриаз. Пароксизмы начинаются утром или после длительного отдыха, провоцируются приемом калия и продолжаются от нескольких дней до нескольких недель.
Содержание натрия и калия в период пароксизма остается нормальным, однако при назначении натрия хлорида состояние больных улучшается.
Заболевание у детей младшего возраста следует дифференцировать от полиомиелита и диэнцефальной эпилепсии.
Лечение должно быть направлено на задержку натрия в организме. С этой целью назначают ацетозоламнд и 9-альфа-фторгидрокортизон, внутривенное введение физиологического раствора.
Профилактика приступов состоит в назначении диеты, богатой солью, и натрийзадерживающих гормонов.
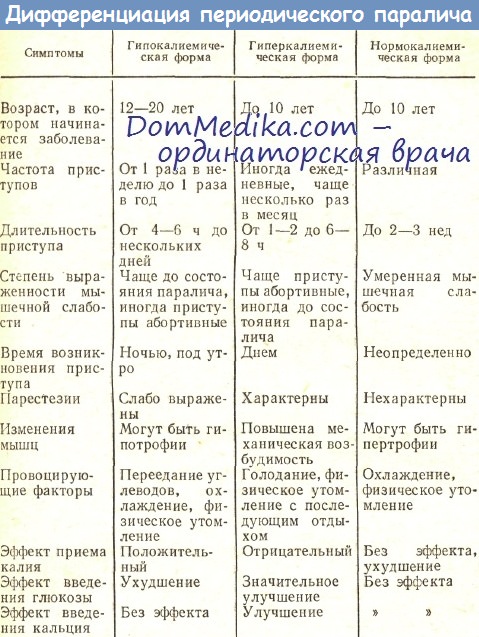
Дифференциально-диагностическая характеристика различных вариантов периодического паралича
Синдром кудрявых волос Менкеса (трихополидистрофия). Наследственное заболевание обмена меди. Описано J. Menkes в 1962 г. Наследуется рецессивно сцепленно с Х-хромосомой.
В основе патогенеза трихополидистрофии лежит дефект всасывания и транспорта меди в кишечнике. В сыворотке крови снижена активность церулоплазмина, уменьшено содержание меди в тканях печени. Дефицит меди в организме приводит к нарушению формирования эластических и коллагеновых волокон, не синтезируются дисульфиды, входящие в состав кератина. Поражение мозга, вероятно, связано с нарушением мозгового кровообращения и с дефицитом цитохромоксидазы (фермента, содержащего медь). В плазме крови повышено содержание глутаминовой кислоты, снижено содержание токоферола.
При патологоанатомическом исследовании в мозговой ткани обнаруживают глиоз, кистозную дегенерацию белого вещества головного мозга. В артериях всего тела выявляются фрагментация и редупликация внутренней эластической оболочки и нарушение расположения эластических волокон среднего слоя. Отмечаются также уменьшение числа ганглиозных клеток в сетчатке глаза и глиальное перерождение зрительных нервов.
Наиболее ранним симптомом является нарушение терморегуляции в виде гипертермии, а затем гипотермии. Дети отказываются от пищи, плохо прибавляют в массе, затем анорексия приводит к тяжелым соматическим нарушениям. Одним из частых проявлений нарушений обмена меди является транзиторная желтуха. Резко выражены нарушения периферического кровотока, на фоне которых развиваются приступы миоклонических судорог. Частые рецидивы судорог приводят к тяжелой мышечной дистонии вплоть до опистотонуса. Очаговая неврологическая симптоматика разнообразна, нередко носит мерцающий характер. Отмечаются спастический тетрапарез, гемипарез, парезы дистальных отделов конечностей, чаще кистей, косоглазие, мелкоразмашистый нистагм, «стеклянные глаза». Снижается зрение, что приводит ко вторичной задержке психического развития.
Волосы у детей в период новорожденности нормальные, однако вновь растущие волосы гипопигментированы, крупно закручены, ломкие. Микроскопическая картина волос обозначается термином pili torti (волосы, закрученные вдоль своей оси).
При рентгенографии выявляется мозаицизм строения костей черепа, метафизарные шпоры бедра, реже плечевой и лучевой костей. Вдоль стволов длинных трубчатых костей может быть субпериостальная кальцификация. Артериографически определяются удлинение и извилистость всех сосудов, неравномерность просвета артерий и их окклюзия.
Заболевание протекает тяжело и в недиагностированных случаях заканчивается смертью у детей в возрасте до 3 лет. Причинами смерти являются острые нарушения мозгового кровообращения и сердечно-сосудистая декомпенсация.
Заболевание дифференцируют от внутричерепной родовой травмы, аномалий развития сосудистой системы, других обменных нарушений и дефектов кишечного всасывания.
Патогенетическая терапия состоит в парентеральном введении меди в дозах, обеспечивающих нормальное содержание микроэлемента в крови. Поддерживающая терапия: 190—220 мг солей меди на 1 кг массы тела в день. Рассчитанную дозу вводят двумя инъекциями в неделю. Проводится симптоматическая терапия, направленная на предотвращение сердечно-сосудистых нарушений и инфекционных заболеваний.
Цель медико-генетического консультирования — диагностика заболевания и его профилактика на основе выявления гетерозиготных женщин и антенатального определения пола плода.