Особую группу медленных инфекций составляют поражения ЦНС, проявляющиеся вакуолизацией серого вещества — так называемым губкообразным перерождением нервных тканей. Ему предшествует развитие первично-дегенеративных процессов при полном отсутствии воспалительных реакций.
Своеобразие патоморфологической картины обусловило первичное название заболеваний — трансмиссивные спонгиоформные [от греч. spongia, губка] энцефалопатии.
Было установлено, что возбудители прионовых инфекций проходят через бактериальные фильтры, не размножаются на искусственных питательных средах, воспроизводят феномен интерференции, что дало основание отнести их к вирусам. Однако американский вирусолог Гайдушек установил, что эти вирусы обладают необычными свойствами, так как они оказались устойчивы к действию многих вирулицидных факторов, а материалом, полученным от погибших животных и людей, невозможно заразить клеточные культуры.
В начале 80-х годов американский биохимик Прузинер установил, что возбудителем скрепи (спонгиоформной энцефалопатии овец и коз) выступает не аномальный вирус, а безнуклеиновый низкомолекулярный белок, названный им инфекционным прионовым белком или прионом. Дальнейшие исследования позволили установить, что прионы вызывают все известные спонгиоформные энцефалопатии. В настоящее время эти заболевания обозначают термином «прионовые инфекции».
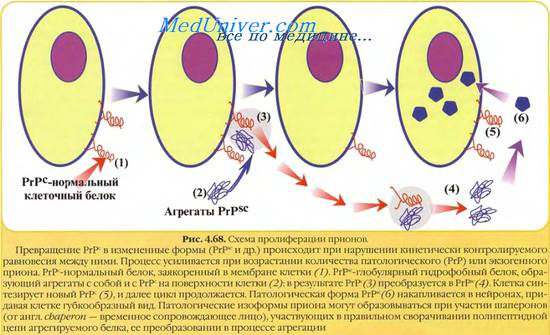
Патогенез поражений прионовых инфекций обусловлен способностью инфекционного прионового белка РrР с (от англ. scrapie, скрепи, являющейся самой распространённой прионовой болезнью) вызывать мутацию гена, кодирующего синтез нормального нейронального прионового белка РrР [от англ. cell, клетка], в результате чего синтезируется инфекционный прионовый белок РrРsс, отличающийся нарушенной пространственной конфигурацией молекулы.
Молекула РrРsс соединяется с молекулой РrРsс с образованием димерного продукта, трансформирующегося в 2 молекулы РrРsс . В следующем цикле 2 молекулы РгР соединяются с 2 молекулами РrРsс, давая начало 4 молекулам PrPSc, что обеспечивает экспоненциальное образование молекул PrPSc. Таким образом, образование инфекционных прионовых белков происходит не за счёт репродукции молекулы РrРsс, попавшей в организм, а за счёт синтеза новых молекул, кодируемых мутировавшим геном РrР .
Физиологическое значение белка РrРsс прионовых инфекций связывают с реализацией функций синапсов, сохранением клеток Пуркинье, регуляцией внутриклеточного содержания Са2+ в нейронах, поддержанием трофики некоторых их популяций и сохранением резистентности нейронов и астроцитов к повреждающим факторам. Белок PrP — короткоживущий (период полураспада 5-6 ч).
В противоположность этому инфекционный прионовый белок PrPSc накапливается в цитоплаз-менных везикулах, что приводит к последующему нарушению функций синапсов и развитию глубоких неврологических дефектов. Позднее PrP с высвобождается во внеклеточное пространство и откладывается в амилоидных бляшках.
У человека прионы вызывают куру, болезнь Кройтцфельдта-Якоба, синдром Гtрстмана-Штраусслера-Шайнкера и фатальную семейную бессонницу. Их характерная особенность — практически полное отсутствие иммунных реакций к инфекционным прионовым белкам, что связано с их внутриклеточной локализацией и структурным сходством с нейрональными белками. Гистологически в тканях мозга выявляют выраженную губчатую дегенерацию, генерализованную гипертрофию астроцитов и амилоидные бляшки, состоящие из белка PrPSc.
Изучение условий возникновения прионовых болезней выявило уникальные особенности их эпидемиологии, отличающие их от прочих инфекционных заболеваний. Они могут формироваться как инфекционные, спорадические и наследственные поражения. В последнем случае предполагают наличие генетической предрасположенности к прионовым инфекционным белкам.