Иммунодефицитный синдром с атаксией-телеангиэктазией. Синдром Луи-Бар
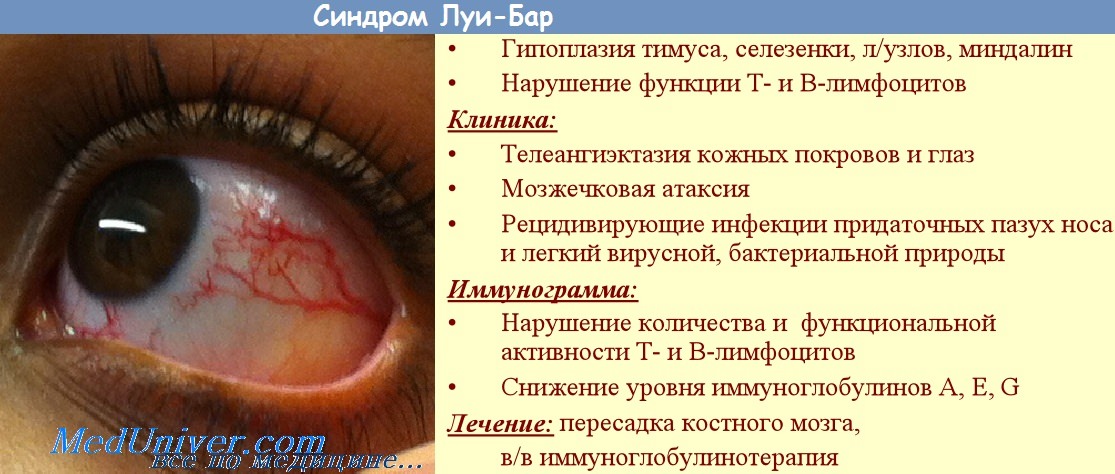
Иммунодефицитный синдром с атаксией-телеангиэктазией [атаксия-телеангиэктазия (AT), синдром Луи-Бар]—наследственный первичный дефицит клеточного и отчасти гуморального иммунитета в сочетании с прогрессирующей мозжечковой атаксией и окулобульбарными телеангиэктазиями.
Этиология синдрома связана с аутосомно-рецессивной наследственностью. R. Peterson и соавт. (1966) высказывали точку зрения, что дефект тимуса, телеангиэктазии и гипоплазия гонад зависят от дефекта мезодермально-мезенхимального зачатка в период эмбриогенеза. Однако этому противоречит вовлечение в процесс ЦНС. Есть точка зрения, что прогрессирующее поражение ЦНС и эндокринных органов обусловлено аутоиммунными процессами, возникающими в результате дефекта тимуса.
W. H. Hitzig (1975) указывает, что у многих больных обнаруживаются в крови аутоантитела. В настоящее время считают, что имеется патогенетическая связь с нарушением функции тимического ретикулоэпителия. Иммунные нарушения зависят от дефекта конечной дифференцировки Т-клеток. Клиника AT характеризуется дефицитом клеточного иммунитета, который, по данным W. H. Hitzig (1975), у 60% больных выражается отсутствием реакции гиперчувствительности замедленного типа и у большинства больных — дефектом лимфоцитов периферической крови, определяемым реакцией с фитогемагглютининами и специфическими антигенами in vitro.
У большинства больных отмечается дефицит IgA, в некоторых случаях описывается, наоборот, увеличение их количества с одновременным уменьшением количества IgG; встречаются также другие варианты дисгаммаглобуминемии. Кроме того, по данным Ю. М. Лопухина, имеет место низкий уровень а-фетопротеина в крови, лимфопения и эозинофилия. Неврологические симптомы болезни вариабельны, у большинства, однако, атаксия развивается с детского возраста в виде нарушений походки, что выявляется обычно до 4 лет жизни. Постепенно атаксия прогрессирует, развивается вторичная мышечная атрофия.
Телеангиэктазии наблюдаются в первый год жизни, иногда позже, на бульбарной конъюнктиве, а затем и в других областях. Больные переживают пубертатный период, но у них не наблюдается развитие вторичных половых признаков. Менструации нерегулярны, у мальчиков прогрессирует атрофия яичек. Отмечается выраженное отставание в физическом развитии. Картину болезни дополняют повторные синопульмональные инфекции. Продолжительность заболевания различна. Иногда больные живут до 39—41 года.
Со стороны лимфоидной ткани обнаруживается значительная гипоплазия преимущественно Т-зависимых зон лимфатических узлов, и селезенки. В солитарных и групповых лимфатических фолликулах желудочно-кишечного тракта отмечается также атрофия с недостаточным формированием фолликулов. В тимусе явления жирового метаморфоза, что не соответствует возрасту.
Дольки представлены очень мелкими островками тимической ткани без телец Гассаля или с единичными образованиями, несколько напоминающими тимические тельца. Кроме того, со стороны эпителия наблюдается появление клеток с гиперхромными ядрами—изменения эти отмечались нами в 2 случаях AT у девочки 10 лет и женщины 24 лет. Со стороны мозжечка — выраженная атрофия с расширением полости IV желудочка.
Микроскопически — тяжелые дистрофические изменения, вплоть до полного исчезновения клеток ганглиозного и зернистого слоя. Подобные изменения отмечают в нейронах передних рогов спинного мозга с демиелинпзациеи задних столбов, в подбугорной области. Со стороны поперечнополосатой мускулатуры вторичная атрофия. В печени очаговые некрозы и диффузная жировая инфильтрация с лимфоплазмоцитарной клеточной реакцией по портальному тракту.
В почках часто встречаются явления хронического пиелонефрита, в легких диффузные бронхоэктазы с абсцессами в сочетании с пневмосклерозом. Наблюдается атрофия яичек и яичников. В передней доле гипофиза дистрофические изменения ацидофильных клеток. Очень характерно сочетание AT с развитием злокачественных опухолей. Чаще встречаются злокачественные лимфомы, лимфогранулематоз, лейкозы, а также медуллобластомы, аденокарциномы, дизгерминомы, ретикулосаркомы.
Мы наблюдали злокачественную неходжкинскую лимфому забрюшинных лимфатических узлов с широкой генерализацией процесса, опухолевой инфильтрацией ткани тимуса у девочки 10 лет. Больные погибают от хронических заболеваний при явлениях общего истощения и выраженного инфантилизма или от злокачественных опухолей, чаще типа гемобластозов.