Гипоталамо-гипофизарная карликовость с глухотой. Мукополисахаридозы: синдром Гурлер
О 2 сестрах с гипоталамо-гипофизарной карликовостью и нейросенсорной глухотой сообщили Winkelmann с сотр.
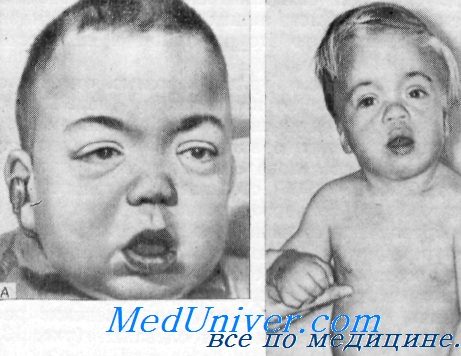
Клинические данные. Данные осмотра. Отмечалась пропорциональная карликовость. При рождении вес и рост были нормальными.
Отставание в росте впервые было замечено при поступлении в школу. У взрослых сестер рост был равен 139 и 146 см.
Эндокринная система. Ни одна из девочек не достигла половой зрелости, ни у той, пи у другой не появились волосы на лобке и не развились молочные железы. У обеих сестер отмечались первичная аменорея и инфантилизм наружных и внутренних гениталий.
Орган слуха. В возрасте около 6—8 лет была замечена быстро прогрессирующая потеря слуха. К 12 годам у обеих сестер отмечалась тотальная глухота.
Вестибулярная система. Результаты исследований не опубликованы.
Лабораторные данные. Рентгенологическое исследование выявило у взрослых сестер общую задержку окостенения (открытые черепные швы и отсутствие синостозирования эпифизов).
Радиоиммунологическое исследование уровня гормона роста в плазме крови показало резкое его снижение (3,0 нг/мл против 30 нг/мл в норме). При стимуляции инсулином уровень гормона роста повышался, но не выше чем до 5,0 нг/мл. Содержание фолликулин-стимулирующего (FSH) и лютеинизируюшего (LH) гормонов соответствовало препубертатному уровню.
Усвоение радиоактивного йода, а также выделение кортизола и кортикостерона были нормальными. Уровень 17-гидрокси- и 11-гидрокси-кортикостероидов в моче был нормальным, даже после стимуляции АКТГ.
Мукополисахаридозы: синдромы Гурлер и Шейе
Мукополисахаридозы — наследственные болезни мукополисахаридного обмена. Дефект активности различных генетически контролируемых путей лизосомалыюй деградации ведет к внутриклеточному накоплению недеградированных кислых мукополисахаридов и к относительно схожим клиническим и скелетным изменениям. Фенотип наиболее резко выражен при синдромах Гурлер и Марото— Лами и менее резко при других мукополисахаридозах (McKusick, Spranger).
В связи с малым объемом этого текста мы сможем только кратко в общих чертах представить клинические и лабораторные данные о тех мукополисахаридозах, которые сопровождаются глухотой.
Синдром Гурлер (МПС-I-Г) является классическим прототипом мукополисахаридозов. Его кардинальными симптомами являются: задержка роста со второго года жизни, выраженная умственная отсталость, прогрессирующее огрубение черт лица, начинающееся примерно в конце первого года жизни, помутнение роговицы, хронические выделения из носа и повторные инфекции верхних дыхательных путей, грыжи, прогрессирующая тугоподвижность суставов, приводящая к деформациям, например, «когтеобразная кисть», гепатоспленомегалия, выраженная соматическая и умственная отсталость, а также биохимические доказательства внутриклеточного накопления и выделения с мочой чрезмерных количеств кислых мукополисахаридов. Смерть обычно наступает до 10-летнего возраста от пневмонии и/или сердечной недостаточности.
При синдроме Шейе (МПС-1-Ш), аллельной форме синдрома Гурлер, черты лица несколько грубоваты, наблюдаются нижнечелюстной прогнатизм и опущенные углы рта. Нос широкий. Начинающееся в молодости прогрессирующее помутнение роговицы приводит обычно к снижению остроты зрения в четвертом десятилетии жизни. Больные слегка отстают по росту от сверстников. Интеллект нормальный. Кисти и стопы широкие, пальцы па руках и ногах фиксированы в «когтеподобном» положении.
Подвижность всех суставов ограничена. Часто наблюдается синдром «туннеля запястья». У большинства больных отмечается регургитация аорты.