Синдром Лемьё — Нимех: синдром Шарко-Мари-Тута, нефрит и глухота
Lemieux и Neemeh описали синдром, выявившийся в двух семьях и характеризующийся начинающейся в детском возрасте прогрессирующей дистальной мышечной атрофией, нефропатией с протеинурией и гематурией, а также прогрессирующей нейросенсорной глухотой. Спорадический случай был описан Hanson с сотр..
Клинические данные. Почки. У всех больных наблюдался нефрит, характеризующийся протеинурией и микроскопической гематурией. Функциональные почечные пробы были нормальными. При биопсии почек обнаружены очаги атрофичных канальцев. У 2 больных были выявлены многочисленные гиалинизированные клубочки. Интерстициальные пенистые клетки были найдены только в одном случае (Lemieux, Neemeh).
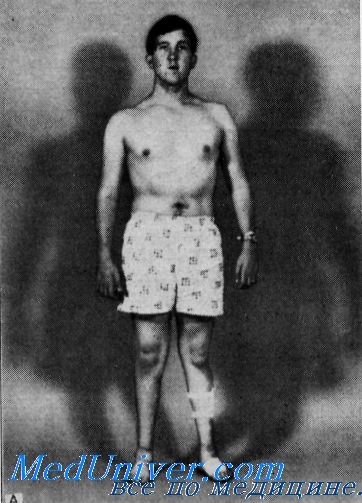
Мышечная система. Дистальная мышечная атрофия начиналась в детстве и медленно прогрессировала. Это состояние сопровождалось прогрессирующей мышечной слабостью, что затрудняло ходьбу и удержание в руках предметов. В старшем детском возрасте медленно развивалась «когтеобразная кисть». Неврологическое исследование обнаружило выраженные атрофии и слабость дистальной мускулатуры ног и собственных мышц кистей. Проксимальные мышцы конечностей оставались нормальными. У больных отмечались неустойчивая походка и двусторонние отвисающие стопы, но истинной атаксии не наблюдалось.
У младшего больного были нормальными рефлексы с двуглавой и трехглавой мышц, а коленные рефлексы и клонус стопы отсутствовали. У 3 старших больных не вызывался ни один из рефлексов. Чувствительность у всех больных была нормальной.
Орган слуха. Нарушения слуха начинались в детстве и медленно прогрессировали. У 2 из 3 больных, изученных Lemieux и Neemeh, а также у больного, описанного Hanson с сотрудниками, была выявлена прогрессирующая нейросенсорная глухота. У последнего больного отмечалась умеренная потеря слуха, выраженная резче на высоких частотах, замеченная впервые в 7-летнсм возрасте. У представленных Lemieux и Neemeh 25- и 34-летних больных отмечалась глухота около 50 дБ. По-видимому, сибсу 21 года аудиометрическое исследование не было проведено.
Вестибулярная система. У мальчика, описанного Hanson с соавторами, с обеих сторон отсутствовала реакция на калорическую пробу.
Лабораторные данные. Скорость проведения в двигательных (локтевом и срединном) нервах составляла около половины нормальной, т. е. от 20 до 25 м/с (Hanson). Электромиография показала отсутствие электрической активности в покое или при попытках добровольного сокращения дистальной мускулатуры. Проксимальные мышцы показали нормальные потенциалы. Полученные данные позволили диагностировать неврогенное происхождение поражения дистальной мускулатуры.
Биопсия икроножного нерва патологии не выявила. Биопсии большой берцовой, икроножной мышц и латеральной широкой мышцы бедра обнаружила изменения, согласующиеся как с миопатией, так и с невральной атрофией.
Наследственность. Наследуется ли этот синдром по рецессивному или по доминантному типу, неясно. Возможно, описанное здесь состояние действительно для двух различных синдромов. В первой изученной Lemieux и Neemeh семье были поражены 4 из 11 сибсов. У одного был синдром Шарко— Мари — Тута, у другого — нефропатия и у двух остальных — оба заболевания. Остальные 7 сибсов и мать не были обследованы. Отец умер от коронарной болезни сердца в 55-летнем возрасте.
Во второй семье у пробанда в возрасте 21 года отмечались дистальные мышечные атрофии и нефропатия, но не было глухоты. У его 12-летней сестры, 13-летнего брата и их 46-летней матери имелись дистальные мышечные атрофии. Исследование мочи выявило у 7 членов семьи легкую протеинурию.
Больной, описанный Hanson с сотр., был единственным пораженным среди 5 братьев и сестер. Его родители были здоровы.
Диагноз. Нефропатия при этом заболевании чрезвычайно напоминает ту, что наблюдается при синдроме Альпорта. Однако синдром Альпорта не сочетается с синдромом Шарко — Мари — Тута.
Лечение. Может оказать пользу применение слуховых аппаратов.
Прогноз. Продолжительность жизни трудно прогнозировать, так как все больные с нефропатией были относительно молодыми. Синдром Шарко — Мари — Тута представляется вполне классическим по своему течению.
Выводы. Синдром характеризуется:
1) вероятным аутосомно-доминантным наследованием;
2) нефропатией, похожей на ту, что наблюдается при синдроме Альпорта;
3) синдромом Шарко — Мари — Тута и
4) прогрессирующей нейросенсорной глухотой, начинающейся в детстве.