Врожденный ангидроз является одной из многих эктодермальных дисплазий. Это состояние может встречаться как изолированный признак, но обычно оно сочетается с другими дефектами кожи и особенно часто с олигодонтией и гипотрихозом. В 1946 г. Helweg-Larsen и Lud-vigsen описали синдром, характеризующийся врожденным ангидрозом и прогрессирующей нейросенсорной глухотой. Авторы лично обследовали 6 больных членов семьи п на основании анамнестических данных диагностировали 8 других случаев.
Клинические данные. Покровная система. У больных наблюдалась неспособность потеть, которая выявлялась уже к концу первого года жизни. При сильном напряжении или при жаркой погоде у них возникало состояние дискомфорта, и они часто не могли работать из-за головной боли, одышки или сердцебиения. На коже, в области подмышек, шеи или переносья вместо пота появлялись кристаллы соли. Крахмально-йодная проба, произведенная одному больному, выявила некоторые доказательства потоотделения только на переносье, под мышками, на предплечьях и в области груди. В каждой из этих областей было обнаружено от 6 до 28 пятен нота.
При мышечной работе число потовых точек не увеличивалось. Пробанд был помещен в жаркую комнату (52С) на 50 мин, а при другой пробе ему ввели подкожно 0,3 мг пилокарпина. Потоотделение у пего было ограничено только несколькими потовыми точками. В противоположность этому, у здоровых лиц из контрольной группы при обоих тестах наблюдалось профузное потоотделение.
Орган слуха. У 5 больных с нарушенным потоотделением была также обнаружена прогрессирующая нейросенсорная глухота, замеченная впервые в 35—40-летнем возрасте. Аудиограммы показали у 2 больных резко выраженную высокочастотную нейросенсорную глухоту. Более подробного описания нарушений слуха не имелось.
Вестибулярная система. О вестибулярных пробах не упоминается.
Лабораторные данные. Общий анализ крови и уринолизис у пробанда были нормальными.
Патология. При биопсии кожи, взятой с предплечья, у 2 больных констатированы нормальные волосяные фолликулы и кровеносные сосуды, но потовые и сальные железы отсутствовали. При биопсии кожи из подмышечной области обнаружено у 1 больного почти полное отсутствие потовых желез. Видна была только одна гипертрофированная потовая железа. Сальные железы отсутствовали.
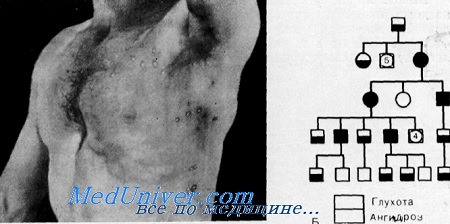
Наследственность. Диагноз ангидроза у 6 обследованных больных был подтвержден. Кроме того, у 8 больных диагноз основывался на анамнестических данных. У 5 больных с ангидрозом отмечалась также прогрессирующая нейросенсорная глухота, которая у 2 больных была подтверждена аудиометрическими исследованиями.
Таким образом, родословная семьи обнаружила 14 больных в пяти поколениях, что соответствует аутосомио-домипантному наследованию.
Диагноз. Описано несколько типов ангидроза. Аутосомно-реиессивно наследующаяся форма, характеризующаяся аигидрозом, без аномалий зубов, лица или мозга, была описана Mahloudji с соавт.. Обе формы (Х-сцспленная и редкая аутосомно-рецессивная) гипогидротической эктодермальной дисплазии включают адоптию и гипотрихоз (Gorlin et al.). Ангидроз часто встречается в сочетании с врожденной сенсорной нейропатией (Vassela et al.). Эти формы наследственного ангидроза отличаются от описанной здесь формы по типу наследования и по сопутствующим аномалиям.
Лечение. Лечение ангидроза симптоматическое. Больные должны воздерживаться от сильного напряжения и не подвергаться действию высоких температур, так как у них имеется субъективная гипертермия. Лечение глухоты заключается в применении слуховых аппаратов.
Прогноз. Глухота, очевидно, прогрессирует медленно. Больных нужно обследовать для точного определения прогрессирования глухоты.
Выводы. Характеристика этого синдрома включает:
1) аутосомно-доминантное наследование;
2) врожденный ангидроз;
3) выявляющуюся в среднем возрасте прогрессирующую нейросенсорную глухоту.