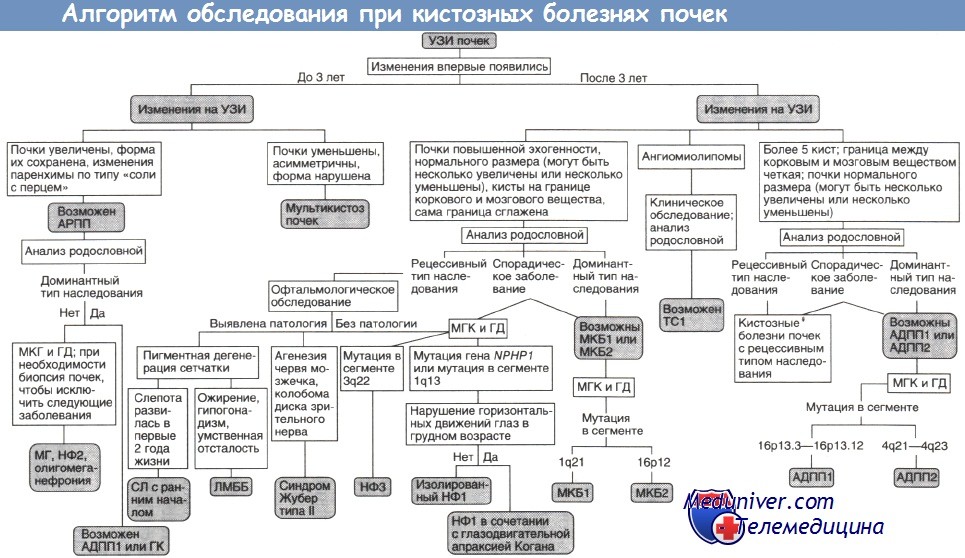
Нефронофтиз и медуллярная кистозная болезнь объединены сходством морфологической и клинической картины, различаясь лишь типом наследования и сроком начала заболевания. В обоих случаях обнаруживают утолщение и разрушение базальных мембран почечных канальцев, лимфоцитарную инфильтрацию интерстициальной ткани, атрофию и расширение дистальных канальцев.
Со временем развивается интерстициальный склероз и формируются кисты — главным образом, на границе коркового и мозгового вещества почек.
В отличие от нефронофтиза медуллярная кистозная болезнь наследуется аутосомно-доминантно, терминальная почечная недостаточность при ней наступает в зрелом возрасте, а внепочечные проявления отсутствуют. Выделяют медуллярную кистозную болезнь почек типов I и II.
В первом случае заболевание связывают с мутацией гена MCKD1, расположенного на 1-й хромосоме (сегмент 1q21). Ген, ответственный за медуллярную кистозную болезнь типа II, находится на 16-й хромосоме (сегмент 16р12) и был локализован при картировании генома с помощью анализа сцепления. У большинства больных медуллярной кистозной болезнью наблюдались гиперурикемия и подагрический артрит.
АДПП1, АДПП2 — аутосомно-доминантныи поликистоз почек типов I и II;
АРПП — аутосомно-рецессивный поликистоз почек;
ГД — генодиагностика (при необходимости ГД включают в МГК);
ГК — гломерулокистоз почек;
ЛМББ — синдромы Лоренса—Муна и Барде—Бидля;
МГ — синдром Меккеля—Грубера;
МГК — медико-генетическое консультирование;
МКБ1, МКБ2 — медуллярная кистозная болезнь типов I и II;
НФ1, НФ2, НФЗ — нефронофтиз типов I, II и III;
СЛ — синдром Сениора—Локен;
TC1 — туберозный склероз типа I.