Диагностика и лечение аутосомно-рецессивного поликистоза почек
Диагноз аутосомно-рецессивного поликистоза почек основывается на клинической картине, данных лучевой диагностики и биопсии почек и печени, а также генодиагностики. При длительном течении заболевания появляется еще один диагностический признак — кистозное расширение внутрипеченочных желчных протоков (болезнь Кароли).
Лучевая диагностика и биопсия аутосомно-рецессивного поликистоза почек
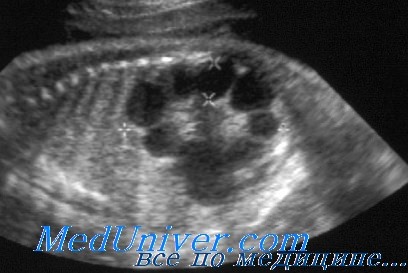
При УЗИ почки резко увеличены, но форма их сохранена. Паренхима имеет повышенную эхогенность, крупнозернистая, с наличием мелких эхонегативных зон («соль с перцем»). На более поздней стадии помимо веретенообразных могут появиться кисты сферической формы. УЗИ печени выявляет повышенную эхогенность, а позднее — хорошо различимые кисты желчных протоков.
Диагностике помогают данные КТ и МРТ почек и печени и экскреторной урографии.
При МРТ собирательные трубочки расширены в виде веретенообразных, радиально расположенных кист, но решающего значения при постановке диагноза эти данные не имеют. В кисты превращены от 10 до 90% собирательных трубочек — в зависимости от тяжести и возраста начала заболевания. При биопсии печени находят разрастание желчных протоков, портальный и перипортальный фиброз.
УЗИ при поликистозе почек
Медико-генетическое консультирование и генодиагностика аутосомно-рецессивного поликистоза почек
Ген, ответственный за аутосомно-рецессивный поликистоз почек, находится на 6-й хромосоме (сегмент 6р21.1—р12). Локусной генетической гетерогенности не выявлено даже для поликистоза с ранним началом (в перинатальном периоде). Так как тип наследования аутосомно-рецессивный, риск заболевания для родных братьев и сестер больного составляет 25%.
В пренатальном периоде диагноз можно заподозрить при маловодий, а затем подтвердить или опровергнуть его с помощью УЗИ. Однако на ранних сроках беременности УЗИ недостаточно информативно, поскольку почки плода могут начать увеличиваться лишь с 24—28 нед внутриутробного развития. Иными словами, отрицательный результат УЗИ, проведенного на ранних сроках беременности, не исключает аутосомно-рецессивный поликистоз почек у плода.
Чтобы отличить аутосомно-рецессивный поликистоз от аутосомно-доминантного с ранним началом, обязательно проводят УЗИ почек родителей больного ребенка. Поскольку сам ген, ответственный за аутосомно-рецессивный поликистоз, не идентифицирован, а известно лишь его положение на 6-й хромосоме, то при наличии хотя бы одного больного в семье для генодиагностики можно воспользоваться анализом гаплотипов.
Дифференциальная диагностика аутосомно-рецессивного поликистоза почек требует исключения других заболеваний, которые проявляются в грудном возрасте и сопровождаются кистами почек. При мультикистозе почки уменьшены, форма их нарушена, характерны пороки развития мочевых путей. Чтобы в столь раннем возрасте проявился аутосомно-доминантный поликистоз, в гене PKD1 должны произойти крупные делении нуклеотидов.
Синдромы Лоренса—Муна и Барде—Бидля легко отличить от аутосомно-рецессивного поликистоза почек по таким дополнительным признакам, как пигментная дегенерация сетчатки, полидактилия, ожирение, умственная отсталость и гипогонадизм. Для синдрома Меккеля—Грубера помимо агенезии или дисгенезии почек характерен широкий спектр внепочечных проявлений, в том числе энцефалоцеле в затылочной области, расщелина неба и пороки развития глаз.
Двустороннюю нефробластому (опухоль Вильмса) отличают от аутосомно-рецессивного поликистоза с помощью лучевой диагностики — опухолевая ткань резко отличается от нормальной паренхимы почек.
Лечение аутосомно-рецессивного поликистоза почек
Тяжелая артериальная гипертония у грудных детей с трудом поддается лечению, но добиться снижения АД чрезвычайно важно, для того чтобы более или менее сохранить функцию почек. Не менее важно своевременное лечение инфекций мочевых путей. Нелегко справиться и с портальной гипертензией. При терминальной почечной недостаточности показаны диализ или трансплантация почки.
Иногда проводят трансплантацию печени — либо одновременно с трансплантацией почки, либо, если показания сохраняются, позднее.
Прогноз аутосомно-рецессивного поликистоза почек
До 1 года доживают 94% мальчиков и 82% девочек, родившихся с аутосомно-рецессивным поликистозом почек. Интересно, что у родных братьев и сестер почечная недостаточность при аутосомно-рецессивном поликистозе почек развивается практически в одном и том же возрасте.
Ген, ответственный за АРПП, расположен на 6-й хромосоме. Но так как точная локализация гена на хромосоме неизвестна, то с помощью ПЦР выявляют группы полиморфных участков ДНК, расположенных рядом с предполагаемым геном АРПП. Такая группа полиморфных участков на каждой из гомологичных хромосом называется гаплотипом. Анализ гаплотипов позволяет установить, из какой гаметы — материнской или отцовской — происходит мутация, и проследить ее передачу потомству, даже если точное положение гена на хромосоме не установлено. В этой семье (родословная изображена в верхней части рисунка) родился мальчик с АРПП. Цель исследования — определить, будет ли болен АРПП и второй ребенок. На рисунке показаны четыре исследованных полиморфных участка ДНК, полученных от четырех членов семьи. Все гаплотипы пронумерованы, а материнские и отцовские хромосомы обозначены разным цветом. Ген АРПП расположен между участками D6S272 и D6S243 хромосомы. Больной мальчик получил по одному гаплотипу, содержащему мутантный ген (указаны крестиками), от матери и от отца. Второй же ребенок в семье получил мутантный ген лишь с отцовской гаметой, в то время как со стороны матери ген не поврежден. Таким образом, этот ребенок будет гетерозиготным носителем гена АРПП и болезнь у него не проявится.