В США и в Европе аутосомно-доминантный поликистоз почек типа I встречается чаще всех других наследственных болезней с аутосомно-доминантным наследованием.
Распространенность этого заболевания составляет 1 на 1000 живых новорожденных, и 10—15% взрослых, находящихся на постоянном диализе, страдают именно аутосомно-доминантным поликистозом. Терминальная почечная недостаточность наступает в возрасте 50—60 лет. Ген PKD1, ответственный за аутосомно-доминантный поликистоз почек, находится на 16-й хромосоме (сегмент 16р13.1) и кодирует недавно открытый белок поликистин 1, в состав которого входят крупный трансмембранный и сложный внеклеточный домены.
Полагают, что поликистин 1 связывается с продуктом гена PKD2 — поликистином 2.
Клиническая картина аутосомно-доминантного поликистоза почек
Аутосомно-доминантный поликистоз почек обычно проявляется лишь в зрелом возрасте. Однако иногда тяжелые клинические проявления, в том числе характерные аномалии лица (лицо Поттер), наблюдаются уже у новорожденного. У детей старшего возраста и у взрослых на аутосомно-доминантный поликистоз могут указывать боль в пояснице, гематурия, частые инфекции мочевых путей, артериальная гипертония.
В брюшной полости пальпируются объемные образования, нередки грыжи брюшной стенки. У взрослых могут возникать аневризмы внутричерепных артерий и дивертикулез кишечника.
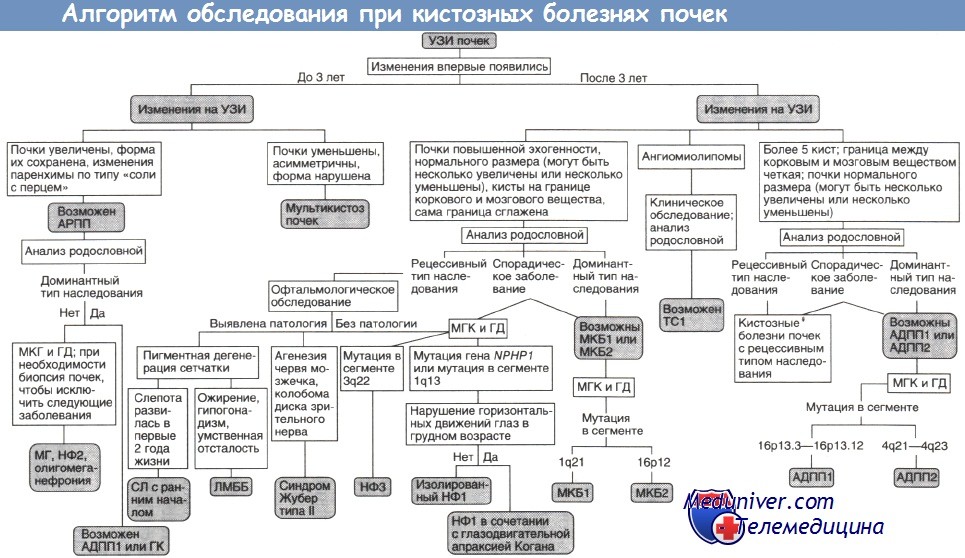
УЗИ почек. У детей с отягощенным семейным анамнезом даже единичная киста при нормальном размере почек может с большой вероятностью оказаться первым проявлением аутосомно-доминантного поликистоза. Помимо почек могут страдать и другие органы, однако до пубертатного периода кисты печени, поджелудочной железы и яичников выявляются редко.
Генодиагностика аутосомно-доминантного поликистоза почек. Несмотря на аутосомно-доминантный тип наследования, длительное течение заболевания с постепенным (за несколько десятилетий) формированием кист предполагает, что в клетках некоторых кист происходят соматические мутации, приводящие к потере гетерозиготности по гену PKD1. Мутации этого гена можно выявить с помощью прямого анализа ДНК.
Лечение аутосомно-доминантного поликистоза почек
Детей из группы риска аутосомно-доминантного поликистоза почек ежегодно обследуют на гематурию, артериальную гипертонию и наличие объемных образований в брюшной полости. Активное лечение артериальной гипертонии и инфекций мочевых путей позволяет замедлить развитие терминальной почечной недостаточности. Для выявления аневризм внутричерепных артерий используют КТ и МРТ. Эти исследования проводят лишь при наличии соответствующей симптоматики или аневризм у родственников больного ребенка.
Аутосомно-доминантный поликистоз почек типа I у грудных детей
Изредка аутосомно-доминантный поликистоз почек проявляется уже в грудном возрасте. Ген PKD1 у таких детей несет крупные делеции, которые могут захватывать и соседний с ним ген TSC2, ответственный за ту-берозный склероз типа II, — так называемый синдром соседних генов. У детей с туберозным склерозом при этом обнаруживаются крупные кисты в обеих почках и первые признаки аутосомно-доминантного по-ликистоза выявляются еще до года.
Аутосомно доминантный поликистоз почек типа II
Это заболевание редко встречается у детей, протекает легче и позднее приводит к терминальной почечной недостаточности — в остальном же его клиническая картина почти неотличима от аутосомно-доминантного поликистоза почек типа I. Ген, ответственный за аутосомно-доминантный поликистоз почек II, идентифицирован, так что отличить один тип поликистоза от другого можно с помощью генодиагностики.
Генодиагностика особенно важна, когда больному аутосомно-доминантным поликистозом планируют провести трансплантацию почки и донором выступает один из членов семьи. Поскольку заболевание проявляется лишь в зрелом возрасте, необходимо убедиться, что предполагаемый донор не несет мутантного аллеля.
АДПП1, АДПП2 — аутосомно-доминантныи поликистоз почек типов I и II;
АРПП — аутосомно-рецессивный поликистоз почек;
ГД — генодиагностика (при необходимости ГД включают в МГК);
ГК — гломерулокистоз почек;
ЛМББ — синдромы Лоренса—Муна и Барде—Бидля;
МГ — синдром Меккеля—Грубера;
МГК — медико-генетическое консультирование;
МКБ1, МКБ2 — медуллярная кистозная болезнь типов I и II;
НФ1, НФ2, НФЗ — нефронофтиз типов I, II и III;
СЛ — синдром Сениора—Локен;
TC1 — туберозный склероз типа I.