Синдромы Прадера-Вилли и Ангельмана. Характеристика
Возможно, наиболее полно изученные примеры роли геномного импринтинга при болезнях человека — синдромы Прадера-Вилли и Ангельмана.
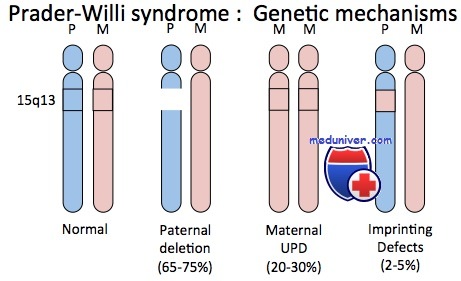
Синдром Прадера-Вилли — сравнительно частый дисморфический синдром, характеризующийся ожирением, чрезмерным и беспорядочным аппетитом, небольшими кистями и стопами, низким ростом, гипогонадизмом и умственной отсталостью. Приблизительно в 70% случаев синдрома наблюдают цитогенетическую делецию, затрагивающую проксимальный отдел длинного плеча хромосомы 15 (15q11-q13), причем только в хромосоме, унаследованной от отца больного.
Таким образом, геном таких пациентов имеет генетическую информацию в области 15q11-q13, происходящую только от матерей. И наоборот, примерно у 70% пациентов с редким синдромом Ангельмана, характеризующегося необычным лицом, низким ростом, выраженным интеллектуальным отставанием, спастикой и судорогами, отмечают делецию приблизительно той же хромосомной области, но теперь в хромосоме, унаследованной от матери; т.е. пациенты с синдромом Ангельмана имеют генетическую информацию в регионе 15q11-q13, происходящую только от отцов.
Эта необычная ситуация удивительным образом доказывает, что родительское происхождение генетического материала в описанных случаях (в хромосоме 15) имеет выраженное влияние на клиническое проявление дефекта.
Приблизительно 30% пациентов с синдромом Прадера-Вилли не имеют цитогенетически обнаруживаемых делеций; но у них выявлены две цитогенетически нормальные хромосомы 15, обе унаследованные от матери. Ситуация иллюстрирует однородительскую дисомию — наличие дисомной линии клеток, содержащих две хромосомы или их части, унаследованные от одного родителя. Если оба экземпляра представлены идентичной хромосомой, состояние называют изодисомией; если присутствуют разные гомологи от одного родителя — гетеродисомией.
Приблизительно 3-5% пациентов с синдромом Ангельмана также имеют однородительскую дисомию, только с двумя неповрежденными хромосомами 15 отцовского происхождения. Эти пациенты служат дополнительным подтверждением того, что синдромы Прадера-Вилли и Ангельмана — результат потери соответственно отцовского или материнского вклада генов участка 15q11-q13.
Кроме хромосомной делеции и однородительской дисомии, несколько пациентов с синдромами Прадера-Вилли и Ангельмана, вероятно, имеют дефект в самом центре импринтинга. В результате не происходит переключения от женского к мужскому импринтингу в сперматогенезе или от мужского к женскому в овогенезе.
Оплодотворение сперматозоидом, несущим аномально персистирующий женский импринтинг, приведет к рождению ребенка с синдромом Прадера-Вилли; оплодотворение яйцеклетки, имеющей несвойственный ей мужской импринтинг, закончится рождением ребенка с синдромом Ангельмана.
Наконец, мутации в материнской копии одного гена — убиквитин-протеин лигазы Е6-АР, как оказалось, вызывают синдром Ангельмана. Ген убиквитин-протеин лигазы Е6-АР расположен в области 15q11-q13 и в норме импринтирован, экспрессируется только материнский аллель в центральной нервной системе (ЦНС).
Полагают, что крупные материнские делеции области 15q11-q13 и отцовские однородительские дисомии хромосомы 15, наблюдаемые при синдроме Ангельмана, служат причиной заболевания, так как приводят к утрате материнской копии критически важного импринтированого гена. Мутации для одного импринтированного гена при синдроме Прадера-Вилли пока еще не обнаружены.