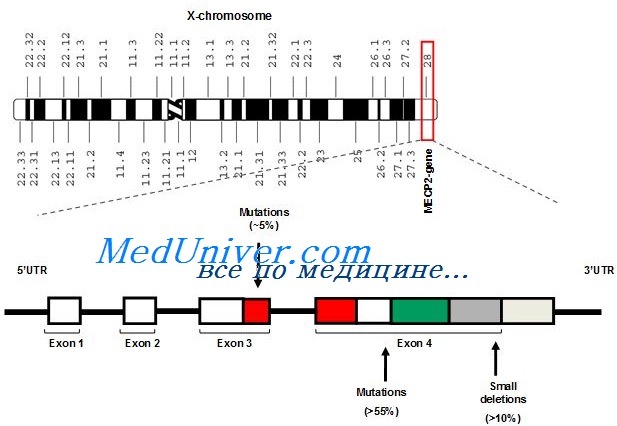
Этиология и встречаемость синдрома Ретта. Синдром Ретта (MIM № 312750) — панэтническое Х-сцепленное доминантное заболевание с распространением среди девочек 1 на 10 000-15 000.
Вызывается мутациями с утратой функции в гене МЕСР2. Описано несколько мальчиков с выраженными нарушениями развития и неврологическими аномалиями с мутациями, вызывающими частичную потерю функции МЕСР2, но обычно у мужчин типичного синдрома Ретта не бывает, кроме случаев кариотипа 47.XXY или соматического мозаицизма.
У нескольких пациентов с атипическим синдромом Ретта найдены мутации в одном, также Х-сцепленном, аллеле гена CDKL5. Белок CDKL5 — киназа треонина и серина, но о его функции мало известно.
Патогенез синдрома Ретта
Ген МЕСР2 кодирует ядерный белок, связанный с метилированием ДНК и переносящий гистоновую деацетилазу в область метилирования ДНК. Точная функция МеСР2 полностью не определена, но существует гипотеза, что он связан с транскрипционным молчанием и эпигенетической регуляцией генов в областях метилированной ДНК. Соответственно дисфункция или утрата МеСР2, наблюдаемая при синдроме Ретта, должна вызывать неправильную активизацию гена.
Мозг у пациентов с синдромом Ретта небольшого размера, с атрофией коры и мозжечка, но без потери нейронов; синдром Ретта, следовательно, не относится к типичным нейродегенеративным заболеваниям. В коре и гиппокампе нейроны пациентов с синдромом Ретта имеют меньшие размеры и более плотно упакованы, чем в норме, и имеют упрощенное ветвление дендритов.
Эти наблюдения указывают, что белок МеСР2 важен для возникновения и поддержки межнейронного взаимодействия, а не для пролиферации предшественников нейронов или их дифференцировки.
Фенотип и развитие синдрома Ретта
Классический синдром Ретта — прогрессирующее неврологическое заболевание, встречаемое почти исключительно у девочек. После периода внешне нормального развития до 6-18-месячного возраста, у больных начинается короткий период замедления и остановки развития с задержкой роста.
Впоследствии они быстро теряют речь и приобретенные двигательные навыки, особенно целенаправленного использования рук. В ходе непрерывного протекания болезни у них развиваются стереотипные движения рук, нерегулярное дыхание, атаксия и судороги.
После краткого периода псевдостабилизации, обычно в дошкольном или ранним школьном возрасте, состояние пациентов вновь ухудшается, появляется выраженная умственная отсталость, прогрессирующая спастичность, ригидность и сколиоз. Больные обычно доживают до взрослого возраста, однако продолжительность жизни уменьшена из-за повышения встречаемости необъяснимой внезапной смерти.
Кроме синдрома Ретта, мутации в гене МЕСР2 вызывают широкий спектр болезней, поражающих как мальчиков, так и девочек. Среди девочек симптоматика колеблется от сильно пораженных пациентов, не способных говорить, поворачиваться, сидеть или ходить, имеющих выраженную эпилепсию, до слабо пораженных пациентов, которые говорят, имеют сохранные двигательные функции, а также сравнительно хорошо сохранившуюся функцию рук.
У мальчиков колебания симптоматики — от внутриутробной гибели, врожденной энцефалопатии до умственной отсталости с различными неврологическими симптомами, или изолированной легкой умственной задержки; классический синдром Ретта описан только у мальчиков с соматическим мозаицизмом по мутации МЕСР2 или с дополнительной Х-хромосомой.
Лечение синдрома Ретта
Заподозренный на основе клинических признаков, диагноз синдрома Ретта обычно подтверждается ДНК-тестированием; тем не менее в настоящее время такое тестирование обнаруживает мутации в гене МЕСР2 только у 80-90% пациентов с типичным синдромом Ретта.
Клинические критерии диагноза для типичного синдрома Ретта включают нормальный пренатальный и перинатальный период, нормальную окружность головы при рождении, сравнительно нормальное развитие до 6-месячного возраста, задержку роста в возрасте между 6 и 48 мес, утрату приобретенных способностей и целенаправленных движений руками к 5-30 мес жизни, и последующее развитие стереотипных движений руками, потерю речевых навыков, выраженную психомоторную отсталость и развитие апраксическои походки и атаксии в возрасте между 12 и 48 мес жизни.
К настоящему времени эффективного лечения синдрома Ретта нет, и помощь сосредоточена на уходе и симптоматическом лечении. Медицинская помощь включает антиконвульсанты при судорогах, прием ингибиторов серотонина, карбидопы или леводопы при ригидности и мелатонина для улучшения сна. Семьи часто нуждаются в социальной поддержке, во взаимодействии с аналогичными семьями через группы взаимопомощи, а в некоторых случаях и в профессиональном консультировании.
Риски наследования синдрома Ретта
Приблизительно 99% случаев синдрома Ретта спорадические; большинство мутаций МЕСР2 возникают вновь, хотя в редких случаях они могут наследоваться от здоровой или мало пораженной матери со смещенной инактивацией Х-хромосомы. По крайней мере, 70% новых мутаций возникают в половых клетках отцов.
Если пара имеет больного ребенка, но мутация в гене МЕСР2 у родителей не выявлена, риск для будущих детей низкий, хотя и выше, чем в общей популяции, из-за возможности необнаруженного полового мозаицизма. Если же мать несет мутацию гена МЕСР2, каждый ребенок, независимо от пола, имеет 50% риск унаследовать мутацию.
Тем не менее недостаточная корреляция между генотипом и фенотипом у пациентов с мутациями в гене МЕСР2 обычно не позволяет давать прогнозы, разовьется ли у женского плода с мутацией МЕСР2 классический синдром Ретта или другая патология. Аналогично, идентификация мутации МЕСР2 у плода мужского пола также не позволяет предсказать внутриутробную гибель, развитие врожденной энцефалопатии или другой патологии.