Этиология и встречаемость семейного полипоза кишечника. По крайней мере, 50% лиц в западных популяциях имеют колоректальные опухоли, включая доброкачественные полипы к возрасту 70 лет, и примерно у 10%, в конечном счете, развивается колоректальный рак. Приблизительно в 15% случаев колоректальный рак имеет семейную форму, включая семейный аденоматозный полипоз (MIM №175100) и ННРК.
Семейный аденоматозный полипоз кишечника (САП) — аутосомно-доминантный синдром предрасположенности к опухолям, вызванный унаследованными мутациями в гене APC. Распространенность семейного аденоматозного полипоза (САП) — 2-3 на 100 000, он вызывает менее чем 1% всех опухолей кишечника. Соматические мутации в гене APC встречаются в более чем 80% спорадических колоректальных опухолей.
Патогенез семейного полипоза кишечника
Белок APC непосредственно или косвенно регулирует транскрипцию, микротубулярный цитоскелет, адгезию, миграцию, апоптоз и пролиферацию клеток. Он формирует комплексы с несколькими другими белками, включая b-катенин.
Для образования аденомы должны инактивироваться оба аллеля APC. Высокая частота соматической утраты функции второго аллеля APC определяет семейный аденоматозный полипоз (САП) как аутосомно-доминантное заболевание. Соматическая утрата функции происходит за счет потери гетерозиготности, внутригенных мутаций, инактивации транскрипции и иногда отрицательного доминантного эффекта унаследованного мутантного аллеля.
Более 95% внутригенных мутаций в гене APC вызывают укорочение молекулы белка APC. Потеря функционального белка APC обычно приводит к высокому уровню в цитозоле свободного бета-катенина; свободный р-катенин мигрирует в ядро, связывается с транскрипционными факторами семейства TCF/LEF и аномально активизирует экспрессию генов. В соответствии с этим механизмом при некоторых колоректальных карциномах обнаруживаются мутации в гене р-катенина без мутаций в гене APC.
Хотя утрата функционирующего белка APC воздействует на клетки, формируя диспластические участки в пределах складок слизистой оболочки кишечника, эти клетки не раковые, и для того чтобы развиться в рак, должны приобрести другие соматические мутации. Этот процесс характеризуется цитогенетической нестабильностью, заканчивающейся утратой больших хромосомных сегментов и, следовательно, потерей гетерозиготности.
Специфические генетические изменения, вовлеченные в этот процесс, включают активизацию онкогенов Ki-ras или N-ras, инактивацию гена-супрессора опухолевого роста в 18q, инактивацию гена ТР53 и изменения в метилировании, ведущем к транскрипционному «молчанию» гена-супрессора опухолевого роста. Поскольку клетки накапливают мутации, они становятся чрезвычайно неопластичны и, в конечном счете, формируют прорастающие и метастатические карциномы.
Фенотип и развитие семейного полипоза кишечника
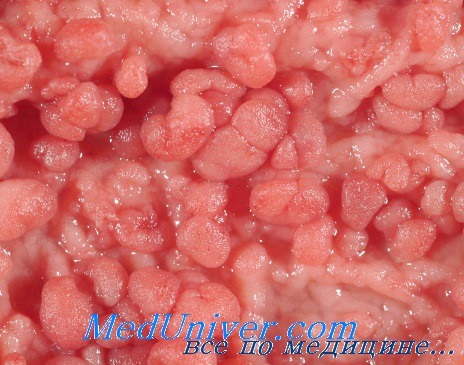
Семейный аденоматозный полипоз кишечника (САП) характеризуется сотнями и тысячами аденома-тозных полипов в толстой кишке.
Клинически заболевание диагностируют по наличию более 100 колоректальных аденоматозных полипов или 10-100 полипов у больного, имеющего родственника с САП. Аденоматозные полипы обычно появляются между 7-40 годами жизни и их количество быстро увеличивается.
При отсутствии лечения колоректальный рак развивается в возрасте 21 года у 7%, к 45 годам — у 87%, а к 50 годам — у 93% пациентов.
Хотя отсутствие пенетрантности бывает очень редко, пациенты с унаследованными мутациями в гене APC не обязательно образуют аденомы или колоректальный рак; они только предрасположены к этому. Этап, ограничивающий частоту образования аденом, — соматическая мутация дикого аллеля гена APC. Перерождение аденомы в карциному требует накопления других генетических изменений. Лица с семейным полипозом кишечника имеют значительно больший риск развития колоректальной карциномы по сравнению с общей популяцией по двух причинам.
Во-первых, хотя среднее время перерождения аденомы в карциному составляет приблизительно 23 года, у таких больных аденомы появляются в гораздо более молодом возрасте, поэтому гибель от других причин до развития карциномы для них менее вероятна. Во-вторых, хотя менее чем 1% аденом переходит в карциному, эти больные имеют десятки тысяч аденом, каждая с потенциалом превращения в карциному. Таким образом, вероятность того, что разовьется, по крайней мере, одна аденома, переродившаяся в аденокарциному, очень высокая.
Пенетрантность и экспрессивность мутаций в гене APC зависят от конкретной мутации гена, генетического фона и среды. Мутации в других регионах гена связывают с синдромом Гарднера (сочетание аденоматозного полипоза толстого кишечника, остеом и опухолей мягких тканей), врожденной гипертрофией пигментного эпителия сетчатки, с легкой формой аденоматозного полипоза толстого кишечника или с синдромом Тюрко (опухоли кишечника и ЦНС, обычно медуллобластомы).
У мышей с мутацией APC количество аденом модифицируют некоторые аллели фосфолипазы А2; аналогичные модификаторы в геноме человека могут вызывать у пациентов с идентичными унаследованными мутациями различные клинические характеристики. Множество исследований спорадического колоректального онкогенеза указывают на повышенный риск для лиц, употребляющих в пищу много животного жира; следовательно, кроме описанного механизма онкогенеза, при семейном аденоматозном полипозе кишечника, вероятно, также играет роль диета.
Особенности фенотипических проявлений семейного полипоза кишечника:
• Возраст начала: от юности до среднего возраста
• Колоректальные аденоматозные полипы
• Колоректальный рак
• Многочисленные первичные опухоли
Лечение семейного полипоза кишечника
Раннее выявление семейного полипоза кишечника необходимо для эффективного вмешательства, т.е. предотвращения колоректального рака. После развития полипов окончательным лечением является тотальная колэктомия с тонкокишечноанальным анастомозом. Рекомендуемое наблюдение для пациентов с риском развития семейного полипоза кишечника — колоноскопия каждые 1-2 года, начиная с возраста 10-12 лет.
Для того чтобы проводить такое наблюдение более целенаправленно, членам семьи больного показано молекулярно-генетическое исследование.
Риски наследования семейного полипоза кишечника
Эмпирический риск колоректального рака в течение всей жизни в западных популяциях составляет 5-6%. Этот риск заметно модифицируется семейным анамнезом. Пациенты, имеющие сибсов с аденоматозными полипами, но без колоректального рака в семейном анамнезе, имеют относительный риск 1,78; относительный риск возрастает до 2,59, если у сибса аденомы развились до 60-летнего возраста. Пациенты, имеющие родственника первой степени родства с колоректальным раком, имеют относительный риск 1,72, который возрастает до 2,75, если колоректальный рак развился у двух или более родственников первой степени родства. Если у больного родственника опухоль развилась до 44 лет, относительный риск равен 5.
В отличие от приведенных данных для всех видов колоректального рака, больные с семейным полипозом кишечника или унаследованной мутацией в гене APC имеют 50% риска родить ребенка с семейным полипозом кишечника при каждой беременности. Отсутствие в семейном анамнезе семейный полипоз кишечника не отвергает этот диагноз у родителя, поскольку приблизительно 20-30% пациентов имеют новую наследуемую мутацию в гене APC. Пренатальная диагностика доступна при анализе сцепления или прямым определением мутации, если она известна у родителей.
Тяжесть, время начала и остальные характеристики не могут быть предсказаны вследствие внутрисемейной вариабельности в экспрессивности.
Наследуемые мутации гена APC не обнаруживают у 10-30% индивидуумов с клиническим фенотипом типичного семейного полипоза кишечника и у 90% индивидуумов со «стертым» семейным полипозом кишечника (фенотип семейного полипоза кишечника с числом аденом менее 100). Среди этих пациентов 10% — гомозиготы или компаундные гетерозиготы по мутациям в ДНК гена репарации MYH; еще 10% передают один мутантный аллель MYH. Гетерозиготность по мутантному аллелю MYH увеличивает риск опухоли кишечника в 3 раза; наличие обоих мутантных аллелей увеличивает риск в 50 раз. Пациента с семейным полипозом кишечника при отсутствии мутаций в гене APC необходимо обследовать на мутации гена MYH, особенно если есть семейный анамнез, напоминающий аутосомно-рецессивное наследование (MIM №608456).
Пример семейного полипоза. Р.П., 35-летний мужчина, направлен в клинику онкогенетики онкологом. Ему выполнена общая колэктомия; слизистая оболочка толстой кишки имела более 2000 полипов и патологические изменения, соответствующие аденоматозному полипозу толстой кишки. Кроме рубцов на животе и колостомы, у больного пигментная аномалия сетчатки — врожденная гипертрофия пигментного эпителия сетчатки. Несколько его родственников умерли от рака. Других проблем со здоровьем в медицинском или семейном анамнезе у него не было. На основе медицинского анамнеза и наводящего на размышления семейного анамнеза генетик заключил, что у пациента наиболее вероятно САП. Генетик рекомендовал схему наблюдения для детей больного и объяснил возможность использования молекулярной диагностики, чтобы узнать риск по САП у детей.
Поскольку пациент не имел контакта с семьей, анализ сцепления был невозможен, и больной решил провести секвенирование гена аденоматозного полипоза кишечника (APC); у него оказалась нонсенс-мутация в 15 экзоне одного из аллелей APC.