Этиология и встречаемость ретинобластомы. Ретинобластома (MIM №180200) — редкая эмбриональная опухоль сетчатки (рис. С-34, см. цв. вклейку), вызванная унаследованными или соматическими мутациями, или их сочетанием, в обоих аллелях гена RB1. Она встречается во всех странах с частотой 1 на 18 000-30 000.
Патогенез ретинобластомы
Белок ретинобластомы — супрессор опухолевого роста, играющий важную роль в регуляции прохождения делящихся клеток через клеточный цикл и выход из него дифференцированных клеток. Белок ретинобластомы выполняет эти две функции, ограничивая активность других факторов транскрипции и усиливая деацетилирование гистонов, модификацию хроматина, связанную с подавлением активности генов.
Мутации в гене RB1, вызывающие ретинобластому, встречаются как в кодирующей области, так и в промоторе гена. Мутации в кодирующей области гена могут дестабилизировать белок ретинобластомы или затруднять ассоциацию с ферментами, необходимыми для деацетилирования гистонов. Мутации в промоторе снижают экспрессию нормального белка ретинобластомы. Оба типа мутаций приводят к недостатку функционального белка ретинобластомы.
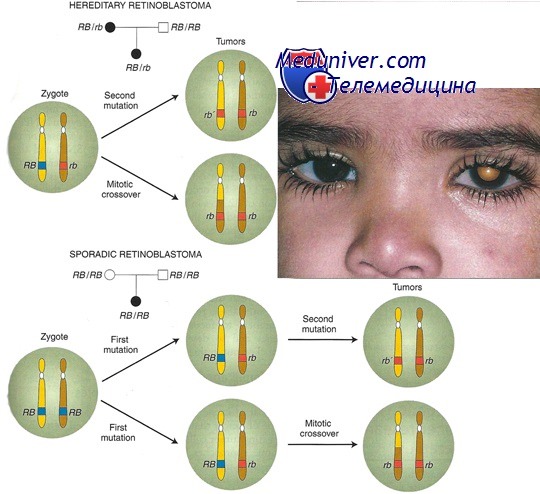
Унаследованную мутацию RB1 обнаруживают у 40% пациентов с ретинобластомой, но только 10% из них имеют других пораженных членов семьи. Мутации RB1 включают цитогенетические аномалии, захватывающие участок хромосомы 13ql4, однонуклеотидные замены и небольшие инсерции или делеции. Существует подтверждение того, что большинство новых унаследованных мутаций возникает в отцовских аллелях, а соматические мутации возникают с равной частотой как в материнских, так и в отцовских аллелях.
Почти половина мутаций происходит в динуклеотидах CpG. При наличии унаследованного аллеля или появления соматической мутации в одном аллеле клетки, для неконтролируемого размножения и развития в ретинобластому также должен утратить функцию и другой аллель RB1 (второй «удар» двухударной гипотезы). Утрата второго функционального аллеля может произойти вследствие новой мутации, потери гетерозиготности или при гиперметилировании участка промотора CpG; чаще всего встречается делеция или изодисомия, реже всего — гиперметилирование промотора.
Ретинобластома обычно передается как аутосомно-доминантный признак с полной пенетрантностью; тем не менее, описано несколько семей с неполной пенетрантностью. Мутации гена RB1, идентифицированные в этих семьях, представляют собой миссенс-мутации, мутации сдвига рамки и мутации промотора. В отличие от более частых аллелей RB1 с полной потерей функции, считают, что эти мутации представляют аллели с некоторой остаточной функцией.
Фенотип и развитие ретинобластомы
Пациенты с двусторонней ретинобластомой обычно проявляются на первом году жизни, тогда как односторонняя форма проявляется несколько позже, с максимумом между 24 и 30 мес. Приблизительно 70% пациентов имеют одностороннюю ретинобластому, а 30% — двустороннюю. Все больные с двусторонней ретинобластомой имеют унаследованные мутации RB1, но не у всех людей с этими мутациями развивается двусторонняя патология. Болезнь диагностируют до 5 лет жизни у 80-95% пациентов.
Ретинобластома однозначно детальна при отсутствии лечения; тем не менее при правильной терапии у 80-90% пациентов удается сдержать болезнь в течение 5 лет после установления диагноза.
Как и следует ожидать при мутациях ключевого регулятора клеточного цикла, пациенты с унаследованными мутациями гена RB1 имеют заметно повышенный риск вторичных неоплазий; этот риск увеличивается влияниями факторов окружающей среды, например от лучевой терапии первичной ретинобластомы. Наиболее частые вторичные опухоли — остеосаркомы, саркомы мягких тканей и меланомы. У пациентов с ненаследственной ретинобластомой увеличения риска злокачественных опухолей нет.
Особенности фенотипических проявлений ретинобластомы:
• Возраст начала: детство
• Лейкокория
• Косоглазие
• Ухудшение зрения
• Конъюнктивиты
Лечение ретинобластомы
Для оптимального исхода важны раннее обнаружение и лечение болезни. Цель терапии — вылечить болезнь и по возможности максимально сохранить зрение. Лечение зависит от размера опухоли и вовлеченности смежных тканей. Варианты лечения при ретинобластоме — энуклеация, различные виды лучевой терапии, криотерапия, лазерная коагуляция и химиотерапия.
Если на момент осмотра болезнь односторонняя, необходимы частые обследования для выявления возможных новых ретинобластом в здоровом глазу, поскольку 30% видимо спорадических случаев вызваны новой унаследованной мутацией. Такие частые обследования обычно продолжают, по крайней мере, до 7 лет жизни.
Для того чтобы более эффективно организовать наблюдение, желательно проведение молекулярных тестов для идентификации мутации в гене RB1. Обычно сначала исследуют образец опухоли, а затем другую ткань, например, кровь, чтобы отличить одну из имеющихся мутаций от унаследованной. Если ни одна из мутаций не унаследованная, такого частого контроля пациенту не нужно.
Риски наследования ретинобластомы
Если у одного из родителей двусторонняя ретинобластома и он, вероятно, несет унаследованную мутацию, эмпирический риск для ребенка равен 45%; это отражает высокую вероятность второй соматической мутации (или «удара») во втором аллеле гена RB1 у ребенка. С другой стороны, если у родителя одностороннее поражение, эмпирический риск заболевания у ребенка составляет от 7% до 15%; это отражает соотношение наследуемых и соматических мутаций у таких пациентов. Почти 90% детей с ретинобластомой — первый случай в семье.
Интересно, что при обследовании сетчатки у 1% здоровых родителей больных детей обнаруживается подтверждение спонтанно разрешившейся ретинобластомы; следовательно, для этих семей риск родить больного ребенка равен 45%. За исключением редкой ситуации, когда один из родителей — непенетрантный носитель мутации гена RB1 семей, в которых ни один из родителей не имел ретинобластомы, риск повторения равен общепопуляционному.
Пример ретинобластомы. Ж.В., 2-летняя девочка, направлена педиатром для обследования по поводу правостороннего косоглазия и лейкокории (отсутствие красного рефлекса зрачка, приводящее к появлению белого зрачка). Ее мать рассказала, что у девочки развилось прогрессирующее правостороннее сходящееся косоглазие на первом месяце жизни, до осмотра педиатра. Она не отметила болезненности, припухания или покраснения правого глаза.
В остальном девочка была здорова. Родители и 4-месячная сестра здоровы; у других членов семьи заболеваний глаз не было. За исключением лейкокории и косоглазия, данные медицинского осмотра оказались нормальными. При офтальмологическом обследовании выявлена одиночная опухоль сетчатки размером 8 диаметров зрительного диска, расположенная около макулы. МРТ головы не обнаружила распространения опухоли за пределы глазного яблока. Ж.В. получила химиотерапию с прицельной лучевой терапией. ДНК-анализ показал, что у девочки присутствует унаследованная мутация (замена С на Т) в одном аллеле гена ретинобластомы (RB1).