Миотоническая дистрофия 1 — аутосомно-доминантное заболевание с наиболее плейотропным фенотипом среди всех болезней экспансии нестабильных повторов. Кроме миотонии, она характеризуется мышечной слабостью и атрофией, нарушениями сердечной проводимости, атрофией яичек, резистентностью к инсулину, катарактами; также существует врожденная форма с умственной отсталостью.
Болезнь вызвана экспансией CTG в 3' UTR гена DMPK, кодирующего белковую киназу. Миотоническая дистрофия 2 — также аутосомно-доминантное заболевание, клинически сходное с миотонической дистрофией 1, за исключением отсутствия врожденного варианта. Миотоническая дистрофия 2 — следствие экспансии тетрануклеотида CCTG в первом интроне гена, кодирующего белок типа «цинковых пальцев».
Поразительно сходные фенотипы миотонической дистрофии 1 и 2 указывают, что у них должен быть общий патогенез. Поскольку нестабильные экспансии происходят в некодирующих участках двух разных генов, кодирующих несвязанные белки, считается, что в основе патогенеза лежит сама экспансия тринуклеотида CUG.
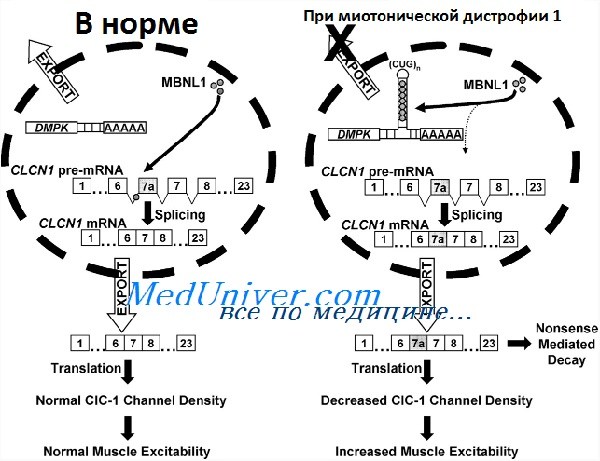
Что же это за механизм, которым большие участки тринуклеотида CUG в некодирующем регионе гена ведут к фенотипам миотонической дистрофии 1 и 2? Оказалось, что патогенез вызван влиянием повторов CUG на РНК-связанные белки.
Следовательно, плейотропия, характерная для болезни, может отражать большое число РНК-связанных белков, на которые влияют повторы триплета CUG. Многие из РНК-связанных белков, подавляемых избыточным числом повторов CUG, — регуляторы сплайсинга, и на самом деле показано, что при миотонической дистрофии 1 нарушения сплайсинга имеют более десяти различных пре-мРНК, включая тропонин Т сердца (объясняющий сердечные аномалии) и рецептор инсулина (объясняющий резистентность к инсулину).
Даже если наши знания аномальных процессов, лежащих в основе миотонической дистрофии 1 и 2, еще неполные, эти молекулярные данные позволяют надеяться, что может быть разработана рациональная низкомолекулярная терапия.