Талассемии, вместе взятые, — наиболее частые моногенные заболевания человека. Это разнородная группа болезней синтеза гемоглобина, при которых мутации уменьшают синтез или устойчивость а- или b-цепей глобина, что вызывает а- или b-талассемию соответственно. Результирующий дисбаланс в соотношении а: bлежит в основе патофизиологических процессов. Цепь, синтезируемая в нормальном количестве, оказывается в относительном избытке; при отсутствии дополнительных цепей для формирования тетрамера избыточные нормальные цепи, в конечном счете, приводят к нарушению клеточной мембраны и вызывают преждевременный распад эритроцитов. Дефект синтеза гемоглобина также приводит к гипохромной микроцитарной анемии.
Название «талассемия» происходит от греческого слова «море», «thalassa», так как болезнь впервые была обнаружена у лиц с берегов Средиземного моря. Как а-, так и b-талассемии, тем не менее, имеют высокую частоту во многих популяциях, хотя a-талассемия — более частая и широко распространенная. Высокая частота талассемии — следствие защитного преимущества у носителей при малярии, аналогично преимуществу гетерозигот у носителей HbS. Существует характерное распределение талассемии в полосе вокруг Старого Света: в Средиземноморье, на Ближнем Востоке, в части Африки, Индии и Азии. В большинстве стран носителей талассемии достаточно много, чтобы стали актуальными проблемы дифференциальной диагностики с железодефицитной анемией и методы обнаружения гомозигот при пренатальной диагностике.
Клинически важно то, что нередко аллели разных типов талассемии и структурных аномалий гемоглобина могут одновременно присутствовать у одного человека. В результате могут происходить клинически значимые взаимодействия разных аллелей одного гена или мутантных аллелей разных генов глобина.
а-Талассемии
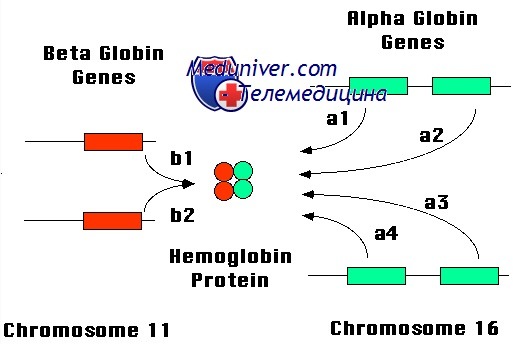
Генетическое нарушение синтеза a-глобина влияет на образование как гемоглобинов плода, так и взрослых и, следовательно, может вызвать как внутриутробную, так и послеродовую патологию. При отсутствии цепей a-глобина образуется избыток цепей из группы b-глобина, формирующих гомотетрамеры гемоглобина. Гемоглобин, содержащий 4 у-цепи, известен как гемоглобин Барта, а тетрамер b4 назван НbН. Поскольку ни один из этих гемоглобинов не способен высвобождать кислород в тканях в нормальных условиях, они не могут выполнять функцию переноса кислорода. Следовательно, младенцы с тяжелой а-талассемией и высоким уровнем гемоглобина Барта имеют тяжелую внутриутробную гипоксию и рождаются с огромным накоплением жидкости, так называемой водянкой плода. При легких формах а-талассемии анемия развивается из-за постепенного осаждения НbН в эритроцитах. Это ведет к образованию включений в зрелых эритроцитах, повреждению клеток и их преждевременной гибели.
Делеции в гене а-глобина при a-талассемии
Наиболее частые формы а-талассемий — результат делеций. Причина высокой частоты этого типа аномалии при мутациях только а, но не b-цепей, была обнаружена при сравнении генов и их локального хромосомного окружения. В каждой хромосоме 16 содержится не просто два идентичных гена а-цепи, но аналогична также и последовательность интронов этих двух генов.
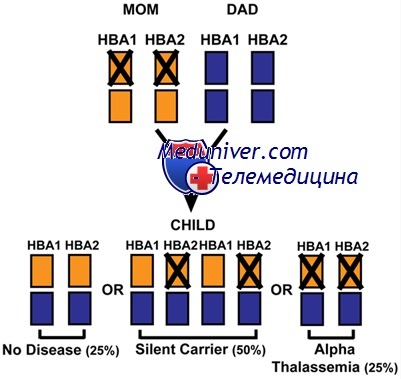
Размещение двойных гомологичных участков как внутри, так и вокруг генов ос-цепей облегчает рассогласование из-за гомологичного спаривания и последующей рекомбинации между областью гена а1 в одной хромосоме и соответствующей областью а2 на другой. Подтверждением того, что такое объяснение происхождения делеций верно, служат сообщения о редких наблюдениях нормальных индивидуумов с утроенным комплексом генов а-цепи. Делеции или другие изменения одного, двух, трех или всех четырех генов вызывают соответствующую тяжесть гематологической аномалии.
Хотя а-талассемии распространены по всему миру, гомозиготные делеции, ведущие к водянке плода, в основном ограничены Юго-Восточной Азией. Высокая частота гена в этой популяции (вплоть до 15% в некоторых регионах) может объясняться природой делеции. Утрата двух генов а-цепей, так называемый «признак» а-талассемии (два нормальных и два мутантных а гена), может встречаться в форме одного из двух генотипов (-а/-а или —/ аа). Последний генотип сравнительно часто бывает у жителей Юго-Восточной Азии, следовательно, потомство может получить две --/-- хромосомы. В других группах признак а-талассемии — обычно результат генотипа -а/-а, когда фактически нет возможности передать фенотип водянки плода.
Кроме мутаций, приводящих к делециям самого гена а-цепи, существуют мутации с утратой только LCR комплекса а-глобина, что также вызывает а-талассемию. Фактически такие делеции впервые указали на существование подобного регуляторного элемента.
Другие формы а-талассемии
Другие формы а-талассемии встречаются значительно реже, чем только что описанные делеции, и имеют меньшее клиническое значение. Тем не менее еще две формы а-талассемии иллюстрируют важные механизмы болезни. В одном случае а-талассемия вызывается мутацией, приводящей к ZF-делеции. Транскрипция РНК, заглушает ген а2-глобина. Во втором случае при синдроме ATR-X а-талассемия и синдромальная умственная отсталость вызваны мутацией в Х-сцепленном гене ATRX, кодирующем хроматин-ремоделирующий белок, необходимый для нормальной экспрессии комплекса а-глобина.
У двух больных из семьи с признаком а-талассемии обнаружена уникальная делеция (названная a-ZF делецией, по инициалам члена семьи, у которого она впервые идентифицирована), с утратой гена а-глобина и последовательности приблизительно 18 килобаз ниже него. Важно, что утраченная последовательность также включала сайт завершения нормальной транскрипции гена LUC7L, расположенного непосредственно около 3'-конца комплекса генов a-глобина, но транскрибируемого с противоположной нити ДНК до гена а-глобина. (Белок LUC7L — широко экспрессируемый компонент U1 — небольшого ядерного рибонуклеинового белкового комплекса, не играющего никакой роли в развитии a-талассемии в этой семье.)
У больных с a-ZF делецией экспрессия гена а2-глобина заблокирована, несмотря на то что сам ген и все локальные и удаленные управляющие цис-элементы остаются целыми. «Молчание» гена а2-глобина — следствие синтеза инвертированной РНК при транскрипции делетированного гена LUC7L, синтез РНК не может нормально завершиться и переходит через точку разрыва a-ZF в отрезок CpG а2-глобина. У носителей делеции a-ZF слияние антисмысловой РНК а2-глобина и РНК гена LUC7L приводит к утрате экспрессии гена а2-глобина в хромосоме с делецией и полному метилированию отрезка CpG а2-глобина.
Патологическая активность антисмысловой a-РНК при делеции ZF сравнима с активностью антисмысловых транскриптов дикого типа, участвующих в нормальном развитии и подавляющих активность генов. Например, показано, что антисмысловые транскрипты CpG участка вызывают метилирование и подавление множества материнских импринтированных генов, а антисмысловые транскрипты XIST, хромосомного локуса инактивации Х-хромосомы, участвуют в ее инактивации. Несомненно, будут найдены и другие примеры патогенных антисмысловых РНК, возникающих вследствие мутаций, так как изучение молекулярных основ болезней продолжается.
Мутации в ATRX, хроматин-ремоделирующем белке при a-талассемии
Во всех ранее описанных классах а-талассемии мутации в генах а-глобина или их циспоследоватпелъностях приводят к уменьшению синтеза а-глобина. Однако один тип а-талассемии активизирует экспрессию гена а-глобина: это синдром ATR-X, вызванный мутациями в гене ATRX, приводящими к нарушению деятельности или экспрессии хроматин-ремоделирующего белка, ATRX, функционирующего в транс-положении.
Вначале синдром ATR-X считали уникальным заболеванием, поскольку наблюдение НbH (b4-тетрамер) в трех семьях на севере Европы было очень необычным случаем а-талассемии у больных европейского происхождения. Кроме того, все больные были мужчинами, также имевшими тяжелую Х-сцепленную задержку умственного развития с большим числом других разнообразных дефектов, включая характерные черты лица, скелетные аномалии и пороки развития мочеполовой системы. Это разнообразие фенотипов указывает, что ген ATRX регулирует экспрессию многих других генов, помимо а-глобинов, но они пока неизвестны.
Хотя точный механизм действия неизвестен, ATRX принадлежит семье хроматин-ремоделирующих белков, обычно функционирующих в больших мультипротеиновых комплексах, изменяющих топологию ДНК. Топологические изменения управляют изменениями состояний нуклеосом. Аномалии метилирования ДНК у пациентов с синдромом ATR-X указывают, что ATRX необходим для поддержания метилирования в определенных областях генома, возможно модулируя доступ метилазы к точкам связывания. Этот факт заслуживает внимания, поскольку мутации в гене, кодирующем другой хроматин-ремоделирующий белок, вызывают синдром Ретта, нарушая эпигенетическое регулирование генов в областях метилирования ДНК, что приводит к неврологической патологии.
Все мутации, идентифицированные до настоящего времени в гене ATRX, — мутации с частичной утратой функции. То, что ген ATRX совершенно необходим для экспрессии а-глобина in vivo, не подтверждается исследованиями пациентов с синдромом ATR-X, имеющих незначительное уменьшение синтезе а-глобина и легкие гематологические дефекты по сравнению с обнаруживаемыми при классических формах а-талассемии. Тем не менее ключевая роль гена ATRX в экспрессии а-глобина подтверждается тем, что пациенты с приобретенным заболеванием — миелодисплазией с а-талассемией имеют соматические мутации в гене ATRX. В наиболее тяжелых случаях синдрома а-талассемии-миелодисплазии мутации останавливают синтез а-цепей почти полностью. Если бы это происходило по причине унаследованной мутации, то привело бы к летальной форме гемоглобинопатии Барта и водянке плода.