Этиология и встречаемость голопрозэнцефалии. Голопрозэнцефалия (МШ №236100) встречается при рождении с частотой 1 на 10 000-12 000 (у девочек в два раза чаще, чем у мальчиков), это наиболее частый врожденный порок мозга у человека.
Голопрозэнцефалия вызвана рядом причин, включая хромосомные и моногенные заболевания, факторы влияния окружающей среды (например, сахарный диабет матери и, возможно, влияние на мать лекAPCтвенных средств, снижающих уровень холестерина — статинов). Заболевание наблюдают изолированно и как часть различных синдромов, например синдрома Смита-Лемли-Опитца.
Несиндромальная семейная голопрозэнцефалия наследуется преимущественно аутосомно-доминантно, хотя описаны случаи аутосомно-рецессивного и Х-сцепленного наследования. Приблизительно 25-50% всех случаев голопрозэнцефалии связаны с хромосомной аномалией; неслучайное распределение хромосомных аномалий указывает на наличие, по крайней мере, 12 разных локусов голопрозэнцефалии, включая 7q36, 13q32, 2р21, 18р11.3 и 21q22.3.
SHH, первый идентифицированный ген с мутациями, вызывающими голопрозэнцефалию, картирован в локусе 7q36. Мутации SHH вызывают приблизительно 30-40% семейных случаев несиндромальной аутосомно-доминантной голопрозэнцефалии и не менее 5% всех случаев несиндромальной голопрозэнцефалии. Другие гены, задействованные при аутосомно-доминантной несиндромальной голопрозэнцефалии — ZIC2 (5%); SIX3 и TGIF (каждый по 1,3%); и РТСН, редко изменяющийся при голопрозэнцефалии.
Патогенез голопрозэнцефалии
Белок SHH — секретируемый сигнальный белок, необходимый для развития, формирующийся как у млекопитающих, так и у насекомых.
Мутации SHH у человека приводят к утрате его функции. Некоторые цитогенетические аномалии, влияющие на экспрессию SHH, относятся к транслокациям, захватывающим 15-256 килобаз в 5'-конце кодирующего участка гена SHH. Эти транслокации называются позиционными мутациями, поскольку они не изменяют кодирующую последовательность, а повреждают отдаленные управляющие элементы или структуру хроматина, изменяя этим экспрессию SHH.
Фенотип и развитие голопрозэнцефалии
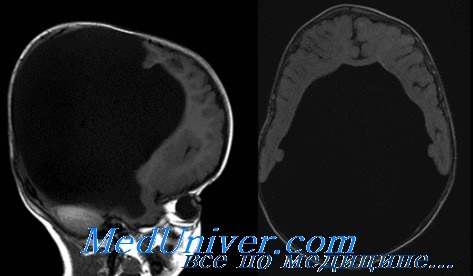
Прозэнцефалические пороки развития при голопрозэнцефалии имеют различную тяжесть, но обычно подразделяются на алобарную голопрозэнцефалию (полное отсутствие межполушарной борозды), полулобарную голопрозэнцефалию (только задний участок межполушарной борозды) и лобарную голопрозэнцефалию (полное разделение желудочков и почти полное разделение коры мозга).
Среди больных голопрозэнцефалией с нормальным кариотипом 63% имеют алобарную, 28% — полулобарную и 9% — лобарную голопрозэнцефалию. Другие частые сопутствующие пороки развития ЦНС — неразделение таламуса, дисгенезия срединной спайки мозга, гипоплазия обонятельных луковиц, гипоплазия зрительного тракта и дисгенезия гипофиза.
Спектр лицевых дисморфии при голопрозэнцефалии варьирует от циклопии до нормы и обычно отражает тяжесть порока развития ЦНС. Дисморфические признаки, связанные, но не патогномоничные для голопрозэнцефалии, включают микро- или макроцефалию, анофтальм или микрофтальм, гипотелоризм или гипертелоризм, дисморфичный нос, аномалии нёба, расщелину язычка, единственный центральный резец и отсутствие уздечки верхней губы.
Задержку развития отмечают почти у всех детей с голопрозэнцефалией. Тяжесть задержки соответствует тяжести пороков развития ЦНС; т.е. пациенты с нормальным мозгом обычно имеют нормальный интеллект. Кроме задержки развития, пациенты часто имеют судорожные припадки, дисфункцию ствола мозга и нарушения сна.
Среди пациентов с голопрозэнцефалией без хромосомных аномалий выживание обратно пропорционально тяжести лицевого фенотипа. Дети с циклопией или этмоцефалией обычно не переживают неделю жизни; приблизительно 50% детей с алобарной голопрозэнцефалией погибает до 4-5 мес, а 80% — до года. Около 50% детей с изолированной полулобарной или лобарной формами голопрозэнцефалии переживают первый год.
Особенности фенотипических проявлений голопрозэнцефалии:
• Возраст начала: пренатальный
• Нарушение развития передних отделов мозга
• Лицевые дисморфии
• Задержка развития
Лечение голопрозэнцефалии
Детям с голопрозэнцефалией необходима быстрая диагностика в первые дни жизни. Лечение — симптоматическое. Помимо медицинских проблем самого больного, основная часть помощи состоит из консультирования и поддержки родителей, а также определения причины голопрозэнцефалии.
Риски наследования голопрозэнцефалии
Этиологически голопрозэнцефалия чрезвычайно разнородна, и риск повторения в семье зависит от основной причины. Матери с сахарным диабетом имеют 1% риска родить ребенка с голопрозэнцефалей. Для родителей пациента с цитогенетической аномалией риск повторения зависит от наличия такой аномалии у одного из них. Для родителей пациентов с синдромальной формой голопрозэнцефалии риск повторения зависит от риска повторения этого синдрома.
При отсутствии голопрозэнцефалии в семье, цитогенетической или синдромальной причины голопрозэнцефалии родителей и сибсов следует тщательно обследовать на наличие стертых форм, микросимптомов, связанных с голопрозэнцефалией, типа отсутствия уздечки или единственного верхнего резца.
Для родителей с отсутствием идентифицированных причин голопрозэнцефалии и стертых малых форм, указывающих на аутосомно-доминантный тип голопрозэнцефалии, эмпирический риск повторения приблизительно составляет от 4 до 5%. В некоторых случаях низкую пенетрантность некоторых мутаций SHH может объяснять дигенное наследование.
Хотя описаны рецессивные и Х-сцепленные формы голопрозэнцефалии, у большинства семей с установленным типом наследования выявляется аутосомно-доминантная форма. Пенетрантность аутосомно-доминантной голопрозэнцефалии приблизительно 70%. Для облигатных носителей аутосомно-доминантной голопрозэнцефалии риск иметь ребенка с выраженной голопрозэнцефалией — 16-21%, а со стертой микроформой — 13-14%. Фенотип носителя никак не влияет на риск иметь больного ребенка и не дает возможности предсказывать тяжесть поражения.
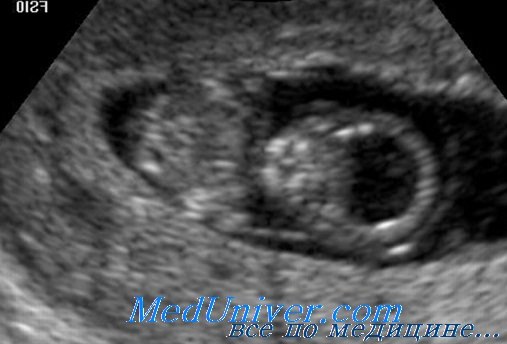
В настоящее время доступно молекулярное тестирование мутаций голопрозэнцефалии. Выраженную голопрозэнцефалию можно обнаружить пренатально при УЗИ на сроке 16-18 нед гестации.
Пример голопрозэнцефалии. Д.Д., 38-летний физик, обратился вместе с женой в генетическую клинику, поскольку их первый ребенок умер при рождении вследствие голопрозэнцефалии. Беременность протекала без осложнений, кариотип ребенка в норме. Ни он, ни жена не указывали на какие-либо проблемы со здоровьем. Сам Д.Д. был приемным ребенком и не знал историю его биологической семьи; семейная история жены не содержала указаний на генетические заболевания.
Тщательное обследование Д.Д. и его жены показало, что у него отсутствовала уздечка верхней губы и отмечался легкий гипотелоризм, других дисморфии не найдено. Врач объяснил, что голопрозэнцефалия у ребенка, с учетом отсутствия уздечки верхней губы и гипотелоризма, напоминает аутосомно-доминантную голопрозэнцефалию. Последующий молекулярный анализ подтвердил, что у отца ребенка есть мутация в гене Sonic Hedgehog (SHH).