Хромосомные аномалии при множественной миеломе - прогноз
Множественная миелома представляет собой клональное новообразование из озлокачествленных плазматических клеток.
Развитию злокачественного процесса нередко предшествует моноклональная гаммапатия; она наблюдается в популяции здоровых взрослых людей с частотой примерно 1 %.
Клоны клеток с хромосомными аномалиями могут быть обнаружены еще до трансформации моноклональной гаммапатии в миелому. При миеломе с помощью стандартного цитогенетического метода клоновые нарушения кариотипа выявляют примерно у 30—50 % больных, причем нередко кариотип резко перестроен.
Применение FISH позволяет обнаружить хромосомные изменения почти у всех больных. Частота этих нарушений нарастает при прогрессировании болезни: до лечения клоны аномальных клеток находят у 18—35 % больных, этот показатель значительно больше у обследованных на стадии III — 40— 60 %, а при плазмобластном лейкозе и появлении экстрамедуллярных опухолей измененный кариотип находят в 85 % случаев.
Идентифицирован ряд неслучайных хромосомных аномалий, характерных для миеломы. Среди них важное место занимают транслокации с участием генов Н-цепей иммуноглобулинов (локус IgH, район 14q32). Связь этих нарушений с развитием болезни и его прогрессией считается доказанной. Видимые под микроскопом изменения локуса IgH наблюдаются в 25—30 % случаев. При молекулярно-генетическом исследовании транслокации этого локуса обнаруживают примерно в 75 % случаев, т. е. в 3 раза чаще, чем при стандартном хромосомном анализе.
Структурные аномалии кариотипа, характерные для миеломы: частота по данным обычного цитогенетического анализа
Аномалия
Частота, %
Перестройки хромосомы 1
30
Перестройки 14q32
25-30
Перестройки 11q13
20
Dcl(13)(ql4)
15-20
8q24
10
Самыми известными партнерами района 14q32 являются следующие участки хромосом: 4р16, 6р21, 11ql3, 16q23; соответственно чаще других выявляются транслокации: t(11;14)(q13;q32), t(4;14)(p16;q32), t(6;14)(p21;q32), t(14;16)(q32;q23). Нередко выявляются различные перестройки 1-й хромосомы, подобные тем, которые обнаруживаются при прогрессировании других гемобластозов и солидных опухолей.
У каждого 5-го больного стандартное цитогенетическое исследование выявляет перестройки длинного плеча хромосомы 13, а применение FISH повышает частоту этой находки до 38—54 %. Характерны также числовые изменения кариотипа: моносомии 2, 13, 14 и 19, а также трисомии 3, 5, 7, 9, 11, 15, 19.
В последние годы выделяют так называемую моносомную миелому, т. е. миелому с гиподиплоидными клонами клеток (клетки с утратой одной или нескольких хромосом). Этот вариант болезни прогностически неблагоприятен.
Остановимся на некоторых молекулярно-генетических изменениях. Известно, что важнейшим результатом неслучайной t(11;14)(q13;q32) является активация гена, ответственного за синтез белка cyclin D1. При t(11;14)(q13;q32) образуется химерный ген IgH/cyclin D1. Это происходит при миеломе в 15— 20 % случаев.
Транслокация (11;14)(q13;q32) на цитогенетических препаратах выглядит абсолютно одинаково при таких разных заболеваниях, как множественная миелома и лимфома мантийной зоны, однако точки разрыва, определяемые молекулярными методами, оказываются разными. При миеломе они локализуются в районе JH или switch, а при лимфоме мантийной зоны — вблизи участков, которые являются мишенями для VDJ-рекомбиназ.
При неслучайной t(6;14)(p21;q32) изменен ген, кодирующий белок cyclin D3. Ген локализован в районе 6р21; эти изменения более редкие, чем изменения циклина D1. Предполагают, что оба гена могут играть важную роль в развитии миеломы.
При характерной t(4;14)(p16;q32) активируются 2 гена — FGRF3 и MMSET, расположенные на небольшом расстоянии друг от друга в коротком плече хромосомы 4(4р16). Эту перестройку невозможно выявить при хромосомном анализе, поскольку при данной транслокации происходит обмен очень маленькими участками хромосом, сходными по окраске, но методы FISH и ПЦР эту транслокацию выявляют.
Ген FGRF3 переносится с хромосомы 4 на хромосому 14 и попадает в область одного из регуляторных элементов (энхансеров) генов IgH, а ген MMSET, оставшийся на хромосоме 4, попадает под влияние другого энхансера, перенесенного с хромосомы 14 на хромосому 4.
Ген FGRF3 кодирует белок с высокой тирозинкиназной активностью. Изменения активности этого гена находят примерно в трети клеточных линий из миеломных клеток, что может свидетельствовать о его существенной роли в злокачественной трансформации этих клеток. Кроме того, ген FGRF3 может быть мишенью для лекарственных препаратов с антитирозинкиназной активностью.
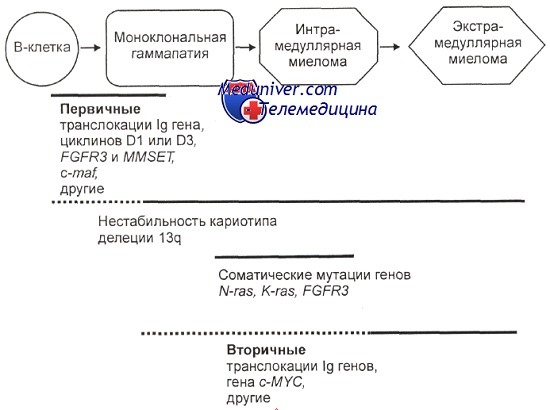
Ступени прогрессии при множественной миеломе: последовательность генетических событий, происходящих на пути от моноклональной гаммапатии до множественной миеломы, а затем и в процессе развития собственно миеломы
Изучение перевиваемых клеточных линий из миеломных клеток позволило установить очень важный факт: каждая линия имеет мутации в одном из следующих генов: FGRF3, K-RAS или H-RAS. Высказывается предположение, что развитие миеломы невозможно без участия одного из трех указанных генов.
Выявлены изменения еще ряда генов: c-MAF (45 % случаев), c-MYC, L-MYC, N-MYC (чаще при прогрессировании заболевания), IRTA1 и IRTA2 (20—30 % случаев) и некоторых других. Следовательно, множественная миелома представляет собой один из многочисленных примеров злокачественной опухоли человека, развитие которой ассоциировано с накоплением множественных приобретенных генетических изменений.
Предполагают последовательность генетических событий в процессе развития миеломы. Постулируется прогрессивное накопление генетических изменений: на сравнительно ранних этапах возникают транслокации с участием района 14q32. Эти изменения, как и делеции хромосомы 13, нередко обнаруживают еще на стадии моноклональной гаммапатии. В результате транслокаций активируются протоонкогены, попадающие в зону влияния регуляторных элементов иммуноглобулиновых генов.
Итогом является иммортализация плазматических клеток. Мутации генов семейства RAS редко находят до развития миеломы, при миеломе их частота составляет около 40 % и нарастает по мере прогрессирования болезни. Для более поздних стадий характерны вторичные транслокации с вовлечением генов семейства MYC, еще позже наблюдается инактивация белков р16, реже — р53. Последние изменения характерны для экстрамедуллярных опухолей.
Изменения кариотипа и прогноз при миеломе (миеломной болезни). В настоящее время ясно клиническое значение далеко не всех хромосомных и молекулярно-генетических нарушений при миеломе, тем не менее некоторые данные уже используются с прогностическими целями. В частности, в группе больных с нормальным кариотипом, установленным при стандартном цитогенетическом анализе, прогноз лучше, чем в группе больных, у которых обнаружены клоны клеток с любыми изменениями кариотипа.
Гипо- или гипердиплоидные клоны прогностически неблагоприятны. При выявлении делеции хромосомы 13, моносомии 13 и делеции длинного плеча хромосомы 11 можно предполагать рефрактерность к терапии. Множественные изменения кариотипа также имеют неблагоприятное прогностическое значение.
У больных без перестроек района 14q32 и изменений хромосомы 13 обычно более благоприятный прогноз, чем у пациентов с перестройками, указанными ранее. Основные показатели группы пациентов с t(11;14) не отличаются от показателей пациентов без этой транслокации. Транслокация (4; 14) и t(14;16) обычно сочетаются с делециями хромосомы 13 и повышенным уровнем b2-микроглобулина. Эта группа прогностически неблагоприятна.
Коррелятивную связь между особенностями кариотипа и эффектом лечения демонстрируют данные В. Barlogie и соавт.. У 49 из 231 больного с миеломой обнаружены аномалии, ассоциированные с плохим прогнозом (11q- и/или 13й). Независимо от типа хромосомных нарушений все больные получили так называемую тотальную терапию с последующей аутотрансплантацией стволовых клеток. У больных с неблагоприятными аномалиями кариотипа бессобытийная выживаемость составила в среднем 25 мес, а у остальных — 52 мес. Похожие результаты получены и другими авторами.
Необходимо подчеркнуть, что хромосомный анализ важно провести до начала лечения для выявления пациентов, нуждающихся в более интенсивной терапии.