Хромосомные аномалии при миелодиспластическом синдроме - прогноз
Кариотип клеток костного мозга у больных с миелодиспластическими синдромами (МДС) интенсивно изучается на протяжении последних 10—15 лет. Аномальные клоны выявлены до лечения у 30—50 % пациентов, в некоторых сообщениях приведены более высокие показатели — 60—75 %.
Обнаружение клонов клеток с аномальным кариотипом при миелодиспластическом синдроме (МДС) имеет важное теоретическое и клиническое значение, поскольку свидетельствует о принадлежности этой группы заболеваний к новообразованиям.
Цитогенетические изменения весьма разнообразны, спектр их близок к спектру хромосомных аномалий, наблюдаемых при остром нелимфобластном лейкозе, особенно вторичных.
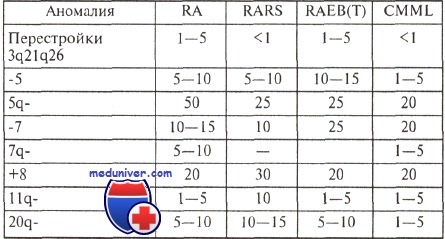
Наиболее характерны моносомии 5 и 7, а также делеции длинного плеча этих хромосом, появление дополнительной хромосомы 8 и делеции длинного плеча хромосомы 20.
Установлено, что частота обнаружения клонов анеуплоидных клеток нарастает по мере прогрессирования болезни: на относительно ранних этапах она составляет 20—30 %, при появлении начальных признаков трансформации в острый лейкоз — до 40—60 %, при трансформации в острый миелобластный лейкоз — 80—90 %.
Транслокации, специфичные для первичных острых нелимфобластных лейкозах, при миелодисплазиях наблюдаются редко. Есть сообщения о повторяющихся транслокациях t(3;3)(q21;q26), t(8;21)(q22;q22) и t(3;21)(q26;q22). Примеры перестроек длинного плеча хромосомы 3 показаны на рисунке.
Частота (в процентах) характерных аномалий кариотипа при различных миелодиспластических синдромах
Основные хромосомные аномалии, характерные для миелодисплазий:
-7 или 7q-
-5 или 5q-
t(1;7)(q10;p10)
del(12)(р12-р13)
t(2;ll)(p13;q23)
del(13)(обязательно с включением 3q14)
t(6;9)(p23;q34)
del(20)(q11ql3)
+8
t(1;3)(p36;q21)
Перечисленные хромосомные аномалии наблюдаются при различных формах миелодисплазий, но частота их несколько различается.
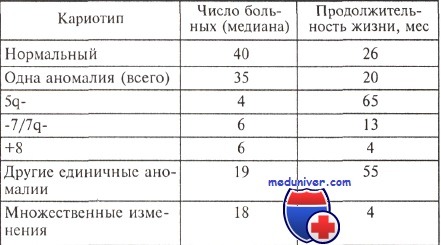
Опыт большинства исследователей свидетельствует о том, что существует корреляция между особенностями кариотипа и продолжительностью жизни больных с миелодиспластическим синдромом (МДС). Относительно благоприятным считается прогноз, если выявлены клоны клеток с единственной перестройкой 5q- или 20q; в то же время при любом варианте миелодиспластического синдрома обнаружение клона с множественными хромосомными аномалиями является крайне неблагоприятным.
Остановимся подробнее на отдельных нарушениях кариотипа, характерных для миелодиспластического синдрома (МДС).
Синдром 5q — рефрактерная сидеробластная анемия у пожилых больных, преимущественно женщин. В новой классификации ВОЗ этот синдром выделен как самостоятельный вариант миелодиспластического синдрома (МДС). Характерна макроцитарная анемия, резистентная к лечению, в костном мозге — признаки миелодисплазии клеток красного ряда и мегакариоцитов. Число тромбоцитов нормально или повышено, в костном мозге наблюдается гиперплазия гиполобулярных микромегакариоцитов. Клиническое течение сравнительно медленное. Трансформации в острый лейкоз наблюдаются приблизительно в 10 % случаев. Синдром впервые описан van den Berghe и соавт. в 1974—1985 гг..
Делеции длинного плеча хромосомы 5 наблюдаются и при других гематологических заболеваниях.
Предполагают, что делетирующийся участок содержит один или более генов-супрессоров. В этом направлении ведутся интенсивные исследования. До настоящего времени не подтверждена важная роль в патогенезе рефрактерной анемии ни одного из изучавшихся кандидатов на роль гена- супрессора.
Продолжительность жизни больных миелодиспластическим синдромом с различными изменениями кариотипа
Синдром моносомии хромосомы 7 встречается преимущественно у мальчиков до 4 лет. Характерна спленомегалия, часто наблюдаются лейкоцитоз с моноцитозом, тромбоцитопения, анемия. Прогноз плохой.
Как отмечалось, утрата одной из хромосом 7-й пары (моносомия 7) наблюдается при самых различных гемобластозах, включая острый нелимфобластный лейкоз, при этом она обычно ассоциирована с неблагоприятным прогнозом.
Делеции короткого плеча хромосомы 17 (17р-) обычно входят в число сложных изменений кариотипа. Как правило, 17р- сочетается с двумя или более хромосомными аномалиями и имеет неблагопрятное прогностическое значение.
В 75 % случаев в присутствии маркера 17р- наблюдается своеобразный дисгранулоцитопоэз в виде псевдопельгеровских гиподольчатых ядер и вакуолизации цитоплазмы. Этот маркер обнаружен не только при миелодисплазиях, но и при самых разнообразных злокачественных новообразованиях, включая солидные опухоли, его присутствие — плохой прогностический признак.
В 1997 г. опубликованы материалы международного совещания, посвященного диагностике и прогнозированию миелодиспластического синдрома. На основании ретроспективной оценки длительности заболевания до перехода в острый лейкоз и общей продолжительности жизни больных результат цитогенетического анализа был расценен как важнейший прогностический признак. В группу с благоприятным прогнозом отнесены случаи с единичными хромосомными аномалия ми: -Y, 5q- и 20q-. Неблагоприятное течение наблюдали при множественных (сложных) нарушениях (три или более перестройки кариотипа) и изменениях хромосомы 7 (делеции длинного плеча, моносомии).
Другие аномалии определяли «промежуточный» прогноз. Средняя длительность заболевания до перехода в острый лейкоз составила по группам 9,4; 0,4 и 1,1—3,3 года соответственно. Эти данные используются при оценке эффективности новых схем лечения миелодисплазии и зарекомендовали себя как одна из лучших прогностических систем при миелодиспластическом синдроме.
Важное диагностическое значение может иметь метод FISH в тех случаях, когда стандартное цитогенетическое исследование неинформативно или обнаруживаются только единичные клетки с нарушением кариотипа, которое по формальным критериям нельзя считать клоном. Для диагностики наиболее характерных хромосомных нарушений при миелодиспластическом синдроме разрабатывается панель FISH-зондов.
Попытки выделить цитогенетические особенности каждой из клинико-морфологических субъединиц, входящих в общую гетерогенную группу миелодиспластических синдромов, не увенчались успехом. В то же время ХММЛ, рассматриваемый как миелопролиферативное заболевание с морфологическими признаками миелодисплазии, нередко ассоциируется со специфической хромосомной аномалией t(5;12)(q33;p13), однако в большинстве случаев ХММЛ эта хромосомная аномалия не выявляется.