Врожденные факторы риска болезней крови - лейкозов, лимфом
Врожденная тенденция нестабильности хромосом или недостаточность системы репараций ДНК относится к безусловным факторам предрасположенности к гемобластозам. Эти виды патологии генома присущи ряду болезней, обусловленных спонтанными разрывами хромосом, а также нерасхождением соматических и половых хромосом на уровне зиготы (синдромы Блума, Фанкони, Тернера, Клайнфельтера, Патау, Дауна и др.).
Частота заболеваний острым лейкозом (ОЛ) и хроническим миелолейкозом (ХМЛ) у детей с такими врожденными аномалиями существенно превосходит фоновую заболеваемость.
Наиболее распространенной врожденной хромосомной патологией является синдром Дауна (СД). В течение 10 лет в Калифорнии (США) было зарегистрировано 1977 больных острым лейкозом (ОЛ) детей до 5 лет. Группу сопоставления составили 1728 контрольных лица. Оказалось, что наиболее высокий риск связан с СД — 22 случая в группе больных острым лейкозом (ОЛ) и ни одного в контрольной. У детей с СД риск возникновения острого лейкоза (ОЛ) в 15—40 раз выше, чем в среднем в популяции (частота 1:100-1:200).
Встречаются как острый лимфобластный лейкоз (ОЛЛ), так и ОНЛЛ, причем определяются два максимума заболеваемости — в младенчестве и в возрасте 3—6 лет. Прогноз заболевания у таких детей крайне неблагоприятный. Редко развивается самоконтролируемая клоновая пролиферация — так называемый преходящий миелопролиферативный синдром. Длительное наблюдение когорты из 4872 больных с СД по материалам регистров Швеции (1965—1993) и Дании (1977—1989) позволило уточнить относительный риск ОЛЛ (п = 22; ОР 24,2; ДИ 95 %: 15,2-36,6) и ОНЛЛ (п = 14; ОР 28,2; ДИ 95 %: 15,7-48,3).
У детей с СД повышен риск также в отношении рака яичка, печени, желудка, болезни Альцгеймера, эпилепсии, ишемической болезни сердца, цереброваскулярных болезней, инфекционных болезней и врожденных аномалий. В последнее время уточнена обратная характеристика частоты: до 10 % детей, заболевших ОНЛЛ (в большинстве случаев это острый мегакариобластный лейкоз), и до 2 % заболевших ОЛЛ страдают синдромом Дауна.
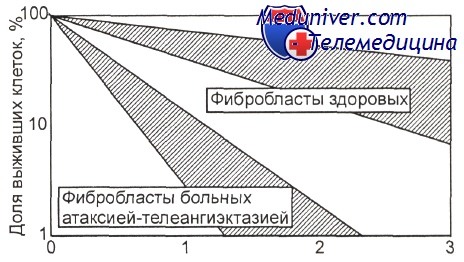
Доля клеток, погибающих при разных дозах у-излучения, в культурах фибробластов, полученных от пациентов с атаксией — телеангиэктазией и от лиц, не страдающих этим заболеванием [Сох R., Masson W. К.]
Анемия Фанкони (АФ) — редкое аутосомно-рецессивное заболевание, сопряженное с врожденными хромосомными аномалиями, прогрессирующей гипоплазией костного мозга и предрасположенностью к новообразованиям. Комплементарный анализ с гибридизацией соматических клеток обнаружил по крайней мере 8 генетически гетерогенных вариантов анемии Фанкони. Очень часто наблюдается моносомия 7. У больных АФ, а также с тяжелой врожденной нейтропенией (синдром Костмана), леченных колониестимулирующим фактором G-CSF, частота острого миелобластного лейкоза (ОМЛ) (или МДС) очень высока (1 изЮ), пик заболеваемости приходится на возраст 14 лет.
У 9 из 145 больных с анемией Фанкони (АФ) при длительном наблюдении зарегистрирован лейкоз, у 18 — другие новообразования. Отношение эмпирического и ожидаемого чисел составило для лейкозов 785:1 (кумулятивная заболеваемость до 48-летнего возраста 10 %), а для прочих новообразований — 48:1.
В отличие от анемии Фанкони, при двух других наследственных аутосомно-рецессивных заболеваниях: синдроме Блума (СБ) и синдроме Луи-Бара, или атаксиителеангиэктазии (AT) — отмечен высокий риск преимущественно острого лимфобластного лейкоза (ОЛЛ). Предрасположенность к лейкозу при СБ связана с повышенной «ломкостью» хромосом и обменами сестринских хроматид, а при AT — дефектностью репараций генных эксцизий. Последнее обстоятельство делает клетки больных AT особо чувствительными к агентам, вызывающим двойные разрывы хромосом.
Это хорошо видно при сравнении дозовой зависимости выживания после радиационного воздействия на культуры фибробластов кожи пациентов с AT и здоровых лиц.
При других врожденных формах гематологической патологии (синдромы Блекфена—Дайемонда, Швахмана, а также тромбоцитопения при неразвитии лучевой кости) риск лейкозов также повышен, но его количественные характеристики не уточнены.
При нейрофиброматозе I типа ориентировочная частота острого миелолейкоза (или ювенильного ХМЛ) 1:200, пик заболеваемости в возрасте 2 года. Часто наблюдается моносомия 7. Похоже, что риск гемобластозов увеличен также при трисомии 18, синдроме XYY и других врожденных состояниях с избыточными хромосомами, однако надежной его оценки пока не получено [53, 210]. Члены семей — носительниц ракового синдрома Ли-Фраумени, который, по всей видимости, обусловлен мутацией ТР53 в стволовой гемопоэтической клетке, также характеризуются повышенным риском лейкозов.
Увеличение риска неходжкиновских лимфом (НХЛ) явно связано с ущербностью иммунитета. Врожденным иммунодефицитом сопровождается СБ, АФ, синдром Клайнфелтера, AT, нейрофиброматоз (болезнь Реклингхаузена) и синдром Уискотта — Олдрича. Среди 176 пациентов с врожденным общим варьирующим иммунодефицитом риск новообразований оказался повышенным, особенно неходжкиновских лимфом (НХЛ) (n = 4; относительный риск 12,1; доверительный интервал 95 %: 3,3—31,0). Отсутствие повышенного риска среди родственников этой группы пациентов свидетельствует о роли именно иммунодефицита, а не других наследуемых генетических факторов.