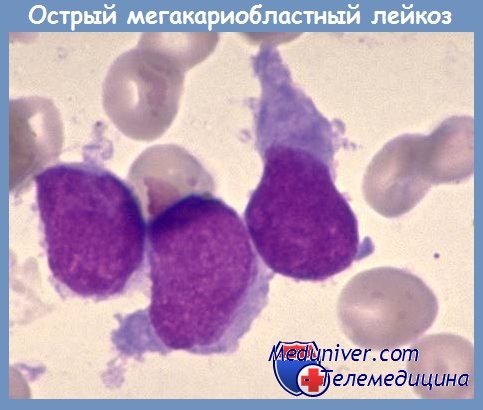
Острый мегакариобластный лейкоз (М7) редкая форма острого миелоидного лейкоза, которую чаще диагностируют у детей первого года жизни, составляет 1—3 % от всех случаев острого миелоидного лейкоза. Для этой формы характерны миелофиброз и остеосклероз, поэтому пунктат костного мозга получить сложно. У детей с М7 могут быть обнаружены литические очаги в костях.
Бластные клетки морфологически могут напоминать таковые при остром лимфобластном лейкозе (вариант L2) или остром миелобластном лейкозе без созревания (M1). Властных клеток может быть немного, но определяются увеличенное количество мегакариоцитов уродливой формы и осколки ядер мегакариоцитов. Точный диагноз может быть установлен лишь при выполнении иммунофенотипирования и соответственно обнаружения характерных маркеров — гликопротеина Ib (CD42; встречается реже), гликопротеина Hb/IIIa (CD41), гликопротеина IIIa (CD61).
В периферической крови чаще всего выявляют панцитопению, тем не менее у многих больных сохранен тромбоцитарный росток: у 30 % пациентов количество тромбоцитов более 100 • 109/л. У детей до 1 года М7 характеризуется гигантской гепатоспленомегалией и нередким определением t(1;22)(p13;q13). В этой транслокации участвуют онкоген N-ras и ген тромбоцитарного ростового фактора бета (PDGFb).
К другим цитогенетическим аномалиям, встречающимся при остром мегакариобластном лейкозе, относятся трисомии 8, 10 и 21, t(1;4), t(3;3), инверсия 3(q21;q26). Прогноз при М7 крайне неблагоприятный.