Дефект Хагемана врожденное расстройство, передающееся аутосомально-рецессивно, которое происходит благодаря недостатку фактора XII и лишено клинического выражения. Первоначально оно было включено в общее расстройство, носящее название гемофилиоидного синдрома, которое в свою очередь отделилось из общего понятия гемофилии.
В 1954 г. Spaet увидел в нем нозологическую сущность, а в 1955 г. Ratnof обособил это расстройство дав ему имя пациента, на котором он его изучил и описал. Оно называется также недостатком контактного фактора.
До настоящего времени цитируются в литературе 140 случаев. Частота этого дефекта оценивается в 0,01/100 000. Не сообщилось ни одного случая приобретенного дефекта Хагемана.
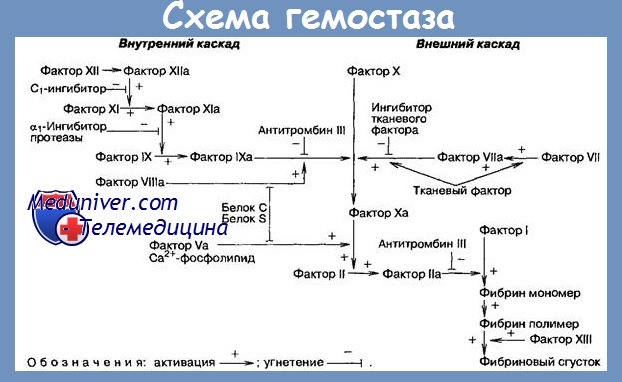
Патофизиология дефекта Хагемана. Основной дефект этого заболевания состоит в ингибиции генов, индуцирующих синтез Ф. XII и помещающихся на соматических хромозомах. В результате происходит недостаточный синтез Ф. XII, что биологически выражается пертурбацией известных лабораторных тестов, но клинически не дает никаких симптомов.
Этот парадокс, кажущийся необъяснимым, повидимому все же имеет свое объяснение: физиологическое значение Ф. XII гораздо больше при коагуляции in vitro чем in vivo, где его удовлетворительно может замещать Ф. XI. (Последний, для активации in vivo нуждается в коллагене и лишь в минимальном количестве Ф. XII; для активации in vitro, он нуждается в гораздо большем количестве Ф. XII, будучи лишенным коллагена).
Генетическая передача — автосомально рецессивная, причем гены имеют малую пенетрацию, что объясняет отсутствие клинического проявления этого заболевания. Встречается в одинаковой пропорции у мужчин и у женщин и не имеет семейного характера. В двух случаях с геморрагическим синдромом мы отметили единокровность родителей.
Клиника дефекта Хагемана. В данном случае нельзя говорить о болезни в клиническом смысле этого слова, так как 97% носителей этого расстройства не страдают геморрагиями; более того, многие подвергались малым и средним хирургическим вмешательствам, не проявляя склонности к геморрагиям. Следует однако отметить, что существовало несколько случаев, которые, вследствие крупных хирургических вмешательств представляли необычайно сильные геморрагии.
Лабораторное исследование для диагностики дефекта Хагемана показывает:
а) Тестыс аномалийными результатами:
1) ВК удлиненный. Характерным является тот факт, что независимо от того, производятся ли тесты в пробирках из простого стекла или в пробирках из силиконового стекла, получаемые время имеют очень близкие значения (в противоположность недостатку Ф. XI).
2) Т.Н., Р.Т.Т. и РТТК постоянно наммного удлинены. Следует отметить, что Р.Т.Т. и РТТК могут корригироваться тем же способом как и при дефекте Розенталя, что указывает на значительное сходство между Ф. XI и Ф. XII. Тест на толерантность к гепарину показывает сильно пониженную способность коагуляции.
3) Т.С.Р. явно сокращен, указывая на недостаточность расхода протромбина.
4) Т.E.G. показывает значительно удлиненные константы „r" и "k" (преобладает „r").
5) T.G.T. дает точно такой же результат, как и при недостатке Ф. XI. Различие между ними делается лишь тестом Носселя с „celite-plasma".
6) Тесты с нормальными результатами: число тромбоцитов, ВК, капиллярная резистентность, ретракция сгустка, T.Q., Т.Ф. II, Т.Ф. V, Т.Ф. VII+X, Т.Ф. VIII, Т.Ф. IX.
Положительный диагноз дефекта Хагемана ставится только на основании лабораторных тестов. Чаще всего он бывает результатом простой случайности, когда речь идет о больных, исследуемых по совсем иному поводу. Диагноз основывается на результатах вышеуказанных лабораторных тестов. Дифференциальная диагностика производится идентичным образом, как и при недостатке Ф. XI. Индивидуализация в отношении дефекта Розенталя делается при помощи теста Носселя с "celite-plasma".
Эволюция дефекта Хагемана самая благоприятная и лишенная осложнений, из всех известных геморрагических диатезов.
Лечение дефекта Хагемана. Практически нельзя говорить о лечении дефекта Гагемана, так как соответствующие пациенты не страдают в клиническом понимании заболевания.
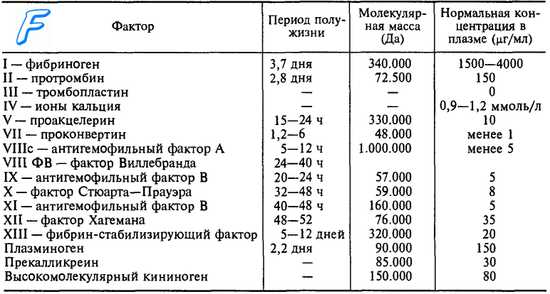
Для чрезвычайно редких случаев, в которых может появляться то или иное геморрагическое явление (вследствие операции или очень тяжелой травмы), мы применяем такое же лечение, как и при дефекте Розенталя, с той единственной разницей, что имея в виду, что продолжительность полужизни Ф. XII 52 часа, а биологический эффект одной перфузии держится 48 часов, порядок введения должен быть по одной перфузии через 2 дня. Продолжительность лечения зависит от эволюции данного случая, обычно 6—10 дней.