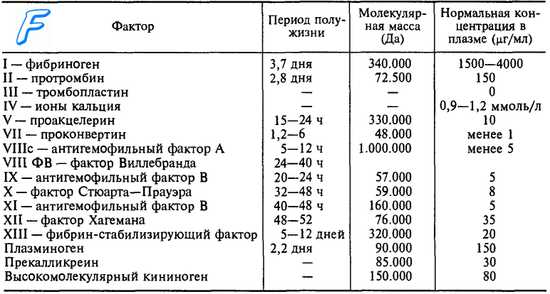
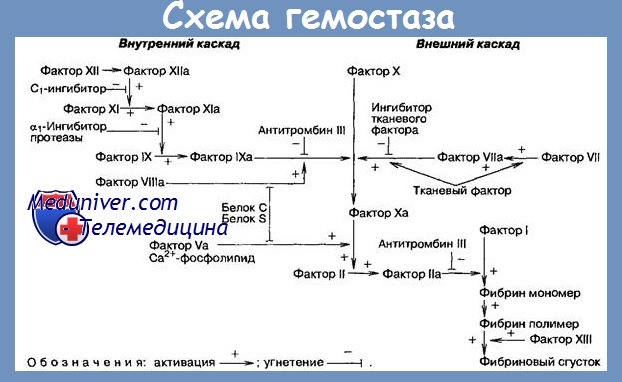
Дефект Стюарт-Прауер - история изучения, причины кровотечений
Дефект Стюарт-Прауер — врожденный геморрагический синдром, возникающий благодаря недостатку синтеза коагуляционного фактора Х и характеризующийся с клинической точки зрения провоцированными, сравнительно тяжелыми геморрагиями. Существует врожденная форма с частично рецессивной автосомальной наследственной передачей и приобретенная, вторичная форма. Первой была обособлена врожденная форма. Вначале, включенная в общую категорию врожденной гипопроконвертинемии, она была открыта в 1956 г. в США совместно Hougie и Graham, которые дали ей название дефекта Стюарт (по имени их пациента), в то время как в Англии, Telfer и Demon назвали ее дефектом Прауер (по имени их пациентки).
В дальнейшем оба названия были присоединены, а конференция в Монтре обозначила недостающий фактор как Ф. X, по предложению Roller и Duckort. В настоящее время этот фактор четко обособлен; он является а-глбулином, в отличие от Ф. VII, который является бета-глобулином.
В литературе опубликовано до сих пор 66 случаев. Частота заболевания довольно низкая — около 0,2/100 000. В подавляющем большинстве случаев врожденная форма является единичной; несмотря на это встречались и ассоциированные недостатки: с Ф. VII (8 случаев), с Ф. IX (4 случая) и с Ф. II, VIII и IX (1 случай).
Приобретенная чистая форма дефекта Стюарта-Прауера отмечена в 5 случаях (4 из них представляли сильный гепатический амилоидоз). Смешанную форму представляют ассоциации с недостатком Ф. II, VII, IX и V, в обстоятельствах, описанных нами в связи с гипопротромбинемией.
Патофизиология дефекта Стюарта-Прауера
Основной дефект врожденной формы состоит в ингибиции генов индуцирующих в гепатоциты синтез Ф. X. Вследствие этого мы наблюдаем у гомозиготов почти тотальный плазматический дефицит Ф. X (0—5%); у гетерозиготов с клиническими признаками, дефицит является средним (10—30%), а у асимптоматических гетерозиготов, легким (40—60%). Имея в виду, что физиологический гемостатический уровень Ф. X — 25%, мы понимаем, почему геморрагический синдром возникает обязательно у всех гомозиготов и факультативно у часть гетерозиготов.
В связи с недостатком Ф. X, новейшие иммунологические исследования определили 2 варианта, связанных с иммунохимической структурой молекулы этого фактора.
— Большинство больных с дефектом Стюарт-Прауер страдают большим или меньшим недостатком Ф. X в плазме; вследствие этого их плазма не обладает иммунологической способностью нейтролизовать антитела анти-Ф. X на пластинках иммунодиффузии. Они были обозначены как CRM; прототипом этой группы был больной Стюарт.
— Меньшинство больных с дефектом Стюарт-Прауер имеют плазму, обладающую способностью нейтрализовать антитела анти-Ф. X и были обозначены как CRM+; прототипом этой группы была больная Прауер. Это значит, что у них в плазме имеется Ф. X с молекулой порочной структуры, которая более не обладает активными коагуляционными свойствами, но сохраняет антигенные свойства и как таковая, может быть обнаружена только иммунологически.
Недавно в рамках группы CRM+ была выделена совсем небольшая подгруппа пациентов, плазмы которых обладают способностью нейтрализовать антитела анти-Ф. X, но лишь в половинной пропорции по сравнению с типичными CRM+; они получили название CRM и считается, что они обладают слабым вариантом антигена.
Наконец, тоже в рамках группы CRM+ была выделена еще одна подгруппа, получившая название F. X, характерная особенность которой состоит в том, что больные имеют сильно удлиненные TQ и РТТК, будучи одновременно и CRM+ (то есть подобными большой классической группе); однако с той разницей, что они имеют нормальный Т. Stypven-цефалнна, а это делает невозможной их дифференциацию (посредством этого теста) от больных с парагемофилией Александера. Дифференциация основывается на РТТК, который в этих случаях удлиненный, в то время как при парагемофилии Александера он нормальный.
Кроме этого, в таких случаях возможно производить детерминацию уровня Ф. X используя параллельно, в качестве реактивов: Стипвен и Тромбопластин. В первом случае, получается очень низкий уровень; во втором случае, получается низкий уровень. Сопоставляя эти результаты с удлиненным РТТК мы подтверждаем диагноз F. X.
Генетическая передача делается автосомально рецессивно, принимая те генотипичные аспекты, которые встречаются при недостатке Ф. VII, но с некоторыми особыми фенотипиями. Ген не полностью рецессивный; он обладает большой пенетрацией и вариабильной выразительностью, что ведет к тому, что этот недостаток представляет наибольшее разнообразие фенотипов. Оба пола затронуты в одинаковой мере. Заболеваемость гетерозиготов намного больше, чем гомозиготов.
Ассоциированная приобретенная форма встречается в тех же этиопатогенетических обстоятельствах как и те, которые мы наблюдаем при недостатке Ф. II.