Болезнь Гоше - синонимы, история изучения, причины
Синонимы болезни Гоше: цереброзидный дислипоидоз, гепатоспленомегалия с липидными клетками типа Гоше, ретикулогистиоцитарный цереброидоз, цереброзиновый липоретикулез.
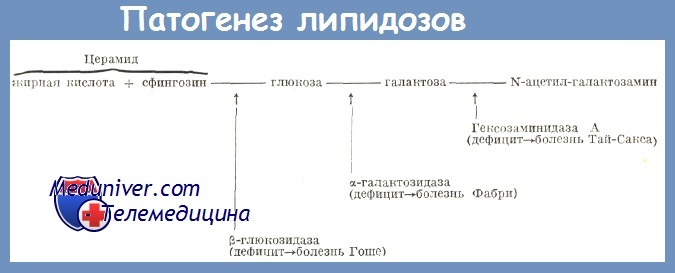
Болезнь Гоше (Б.Г.) — семейный сфинголипидоз, основное расстройство которой состоит в врожденном отсутствии или понижении b-глюкозидазы, которое влечет за собой накопление глукоцеребозидов в макрофагах селезенки, печени, костного мозга, лимфатических желез, нервной системы и других тканей. Способ передачи — аутосомально рецессивный.
Болезнь описал впервые французский дерматолог Gaucher в 1882 г., который считал ее неопластической природы (эпителиома селезенки). В 1900 г. Bovaird отметил семейный характер и гиперпластическую природу болезни. Schlanghaufer (1906) определил болезнь Гоше как системную болезнь лимфогематопоэтического аппарата.
Годом позже, Marchand доказал, что образование клеток Гоше — результат отложения чужого вещества, которое Ebstein и Lieb (1924) идентифицировали как керазин. В дальнейшем утверждалось, что накопление цереброзидов не является результатом повышенного плазматического подвоза, а метаболического внутриклеточного дефекта (Tannhauser).
Изучение энзиматических этапов гликолипидного метаболизма установило, что болезнь Гоше является сфинголипидозом, а дефицитный энзим b-глюкозидаза (Brady). Генез клеточных включений был уточнен впоследствии при помощи исследований клеток Гоше на электронном микроскопе (Neimann, Metayer).
Этиология болезни Гоше неизвестна; частота — редкая (1 случай на 24 000 интернирований). Болезнь появляется в раннем детстве, у детей старшего возраста и даже у молодых взрослых. Около 50% случаев диагностицируются в первые 10 лет жизни. Оба пола затронуты в одинаковой мере. Несмотря на то, что частота явно выше у ашкеназиевых евреев, болезнь была описана во всех расах.
Наследственный характер болезни Гоше был установлен с достоверностью, так как она часто встречается у братьев и реже у последовательных поколений. Способ передачи — автосомально рецессивного типа. В случаях, возникших у последовательных поколений, допускается передача автосомально доминирующего типа, с вариабильной пенетрацией (Kolodny). Выражалось мнение, что болезнь Гоше может появляться и вследствие спорадических мутаций, после чего она передается главным образом по доминирующему принципу.
Патогенез болезни Гоше. Основной генетически детерминированный биохимический дефект болезни Гоше состоит в отсутствии или явном сокращении b-глюкозидазы. Расстройства наступают в следующем порядке: дефицит b-глюкозидазы => аберрантный метаболизм цереброзидов=>тезауризация=>образование клеток Гоше.
У всех больных b-глюкозидаза оказалась пониженной, но в вариабильных размерах, что указывает на существование нескольких типов болезни Гоше (Beutler, Brady, Nitowsky, Turpin). У пациентов с детской формой активность глюкоцеребрози-дазы в тканях сильно понижена по сравнению с нормой (между 0 и 9%) и главное поражение относится к нервной системе. При взрослой форме эта активность колеблется между 12 и 44% (Brady).
У носителей, активность b-глюкозидазы находится на интермедиарных уровнях, между нормальным и существующим у пациентов с выраженной болезнью (Beutler).