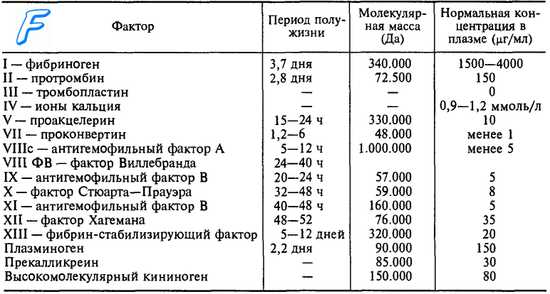
Болезнь фон Виллебранда - история изучения, причины
Болезнь фон Виллебранда наследственный геморрагический синдром передающийся аутосомально доминантно и характеризующийся с клинической точки зрения геморрагиями капиллярного типа в сочетании с умеренными явлениями гемофилиоидного типа. Возникает на основе полного недостатка Ф. VIII, то есть: Ф. VIII—С, Ф. VIII—Ag, и Ф. VIII—vW.
Как нозологическая сущность, болезнь известна с 1926—1931 гг., когда ее открыл и описал von Willebrand. Она носила разные названия: ангиогемофилия, сосудистая гемофилия, тромбопатия Виллебранда-Юргенса; Alexander (1953) и Nilsson (1957) дали ей настоящее название, причем болезнь была окончательно включена в ряд геморрагических плазматических диатезов.
Очень многие работы показали, что тромбоциты больных с болезнью фон Виллебранда (BvW) не функционируют нормально. Посредством тестов на адгезивность Salzman показал, что это свойство тромбоцитов глубоко альтерировано у больных с болезнью фон Виллебранда, при которой тромбоциты очень мало или совсем не задерживаются колонной микрожемчущин. В тестах на тромбоцитарную аггрегацию на ристоценин, тромбоциты нормального лица и гемофилика А отвечают положительно; тромбоциты больных с болезнью фон Виллебранда отвечают отрицательно. Зато, тромбоцитарная аггрегация на адреналин и ADP положительная, как у нормальных лиц, так и у гемофиликов и больных болезнью фон Виллебранда. Эти факты привели к выводу, что тромбоциты при болезни фон Виллебранда имеют нормальное поведение в взаимоотношениях между собой, так как аномалийность появляется только в случае прилипания к субэндотелиальным слоям. Таким образом был выдвинут постулат о существовании плазматического фактора (F.vW), необходимого для тромбоцитариой деятельности адгезивности и аггрегации на ристоцетин. Новейшие исследования доказали правильность этой гипотезы, идентифицируя этот фактор в качестве одной из субфракций молекулы Ф. VIII (Ф—VIII—vW).
С другой стороны, в результате параллельного коагулологического и иммунологического исследования нормальных лиц, гемофиликов А и больных с болезнью фон Виллебранда было установлено следующее: абсолютно все нормальные лица имеют в плазме Ф. VIII—С и Ф. VIII—Ag на высоких уровнях (50—150 ед/100 мл); абсолютно все гемофилики А имеют Ф. VIII—С на очень низком уровне, но Ф. VIII—Ag в норме; у больных с болезнью фон Виллебранда и уровень Ф. VIII—С и уровень Ф. VIII—Ag — понижены.
Сопоставив результаты этих 2 серий исследований, авторы пришли к общепризнанному заключению, что при болезни фон Виллебранда имеет место абсолютный дефицит Ф. VIII, то есть всех трех составных фракций (С, Ag, vW). Следовательно аномалийность Ф. VIII при болезни фон Виллебранда намного сложнее, чем при гемофилии А, где она касается только одной фракции молекулы Ф. VIII (фр. С).
Болезнь фон Виллебранда происходит следовательно на почве тройного врожденного недостатка плазматических факторов, передающегося по наследству автосомально доминантно. Этим объясняется, почему она появляется с одинаковой частотой и у мужчин и у женщин. Впрочем наибольшая часть редких случаев гемофилии у женщин оказалась, при более тщательном анализе, в действительности болезнью вон Виллебранда.
Относительно механизма возникновения болезни существуют 2 теории: мономолекулярная и тримолекулярная. Мономолекулярная теория утверждает, что Ф. VIII является единой молекулой (VIII—Ag), которая имеет на каждом из двух концов по одному ситусу, что и детерминирует ее 2 фракции: VIII—С и VIII—vW. Гемофилия А происходит на почве аномалийности детерминирующего ситуса для VIII—С, причем остальная часть молекулы остается нормальной. Болезнь фон Виллебранда происходит якобы из-за недостатка синтеза всей молекулы. Эта теория более давняя и на ее место пришла более новая тримолекулярная теория. Согласно последней, все 3 фракции Ф. VIII синтетизируются независимо одна от другой, по разным кодомам и лишь в циркуляции они связываются в единый гигантский комплекс. Местами синтеза повидимому являются: для Ф. VIII—Ag и Ф. VIII—vW—клетки сосудистого эндотелия, и для Ф. VIII—С — гепатические клетки. При гемофилии, недостаток синтеза касается только молекулы ф. VIII—С; при болезни фон Виллебранда — всех трех молекул: двух непосредственно (VIII—Ag и VIII— vW) и одной косвенно (VIII—С). Сопоставляя таким образом все новейшие сведения, полученные в этой области, можно с уверенностью утверждать, что на внутреннем генетическом уровне механизм расстройства при BvW повидимому действует по гораздо более сложной схеме, чем при гемофилии.
Порок касается двух пар генов, которые помещаются на тех же соматических хромосомах в смежных локусах. Одна из пар синтетизирует плазматический фактор тромбоцитарной адгезивности (Ф. Виллебранда-Нилссона, Ф. антикровотечения, Ф. VIII—vW). При болезни фон Виллебранда они ингибиро-ваны и более не синтетизируют Ф. VIII—vW. Отсутствие этого фактора ведет к тому, что тромбоциты не прирастают к коллагену и препятствуют таким образом нормальному развитию механизма первичного гемостаза, что имеет своим результатом удлинение ВК (капиллярный геморрагический синдром).
Вторая пара генов является индукторами производства Ф. VIII—Ag. Эта протеиновая молекула — нормальный биологический стимул генов, регулирующих производство Ф. VIII—С, которые также помещаются на паре соматических хромосомов. Они заказывают генам с хромосомов «X» уровень синтеза Ф. VIII—С. Однако благодаря тому, что гены, производящие Ф. VIII—Ag, оказываются ингибированными при BvW, последний синтетизируется мало или совсем не синтетизируется. Это отражается на генах, регулирующих уровень Ф. VIII—С, которые не получая стимула, не отдают команду на производство генам, синтезирующим Ф. VIII—С, вследствие чего последний производится в очень малом количестве. Следовательно, здесь ответственность несут не гены, производящие Ф. VIII—С, как при гемофилии (прямой эффект), а отсутствие стимулирования регулирующих генов (косвенный эффект).
Низкий уровень Ф. VIII—С имеет своим результатом появление гемофилиоидного геморрагического синдрома, более умеренного, чем гемофилический, так как недостаток Ф. VIII—С не падает до таких низких уровней, как при гемофилиях (ни уровень Ф. VIII—С у того же больного не такой стабильный как при гемофилии).
Дифференциация между болезнью фон Виллебранда и гемофилией стала возможной вначале на основании изучения эффекта их перекрестных трансфузий. Трансфузия свежей нормальной плазмы корригирует дефекты обоих заболеваний. Трансфузия тромбоцитов не корригирует ни одного. Трансфузия плазмы от гемофилика А корригирует ВК больного с болезнью фон Виллебранда, а спустя несколько часов начинает подымать его уровень Ф. VIII—С, так что такая трансфузия корригирует до следующего дня и его гемофилиоидное расстройство (объяснение: трансфузирован Ф. VIII—vW и Ф. VIII—Ag. стимулирующий синтез Ф. VIII—С; у гемофилика имеется стимулирующий Ф. VIII—Ag, но отсутствует способность синтеза, а больной с болезнью фон Виллебранда имеет способность синтеза, но ему недостает стимулирующего Ф. VIII—С. Трансфузия плазмы от больного с болезнью фон Виллебранда гемофилику не имеет никакого эффекта (объяснение: больной с болезнью фон Виллебранда не может передавать через трансфузию больному гемофилику генетический кодекс синтеза Ф. VIII—С, который у последнего отсутствует!).
Тот факт, что дефективные гены помещаются на соматических хромосомах объясняет независимость болезни от пола, а доминирующий характер порока объясняет прямую передачу от любых родителей своим потомкам. Следует подчеркнуть, что у гетерозиготов болезнь представляет более легкую клиническую форму; гомозиготы (очень редкие, за исключением жителей Ааландских островов) представляют очень суровые клинические формы.
Частота заболеваемости — 10/100 000, как и при гемофилии. За последние годы было сообщено и 7 случаев приобретенной болезни фон Виллебранда. В этиопатогенезе этих случаев, фигурирует как первичное заболевание: диссеминированная красная волчанка (6 случаев) и моноклональная гаммапатия IgG (1 случай). Все указывают на существование иммунологического механизма.