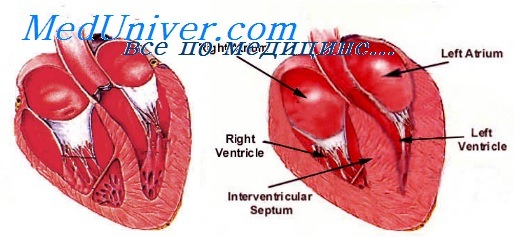
Причины нарушения ритма при дефектах перегородок сердца. Двухстворчатый аортальный клапан
К электрофизиологическим нарушениям, обусловленным мутацией гена NKX2-5, относят прогрессирующее замедление атриовептрикулярпого проведения с блокадой типа Wenckenbach второй степени, слабостью синусового узла и ФП. У некоторых пациентов нарушения электрофизиологической функции сердца наблюдаются в отсутствие его морфологических дефектов.
Замедление АВ-проведения развивается бессимптомно и неизбежно влечет за собой необходимость имплантации постоянного водителя ритма даже через десятилетня после определения структурных дефектов и предполагает постоянное длительное наблюдение за такими пациентами. При обнаружении нарушений на ЭКГ у больного с вероятным спорадическим ДМПП необходимо тщательно изучить семейный анамнез и обследовать близких родственников.
Ген NKX2-5 млекопитающих, гомолог гена мушки Drosopbila tinman (называемый так из-за делеций недоразвития дорсального сосуда, аналога сердца у насекомых), локализован на хромосоме 5q34. Ген NKX2-5 принадлежит к семейству NK-генов, кодирующих факторы транскрипции с гомеобоксмотивом и играющих существенную роль в дифференцировке сердечных прогениторных клеток из мезодермы в клетки млекопитающих и других животных. Организмы-модели с отсутствием тепа NKX2-5 погибают на ранней стадии развития из-за невозможности формирования сердца.
Экспрессия некоторых генов-мишеней, активность которых у мышей регулируется геном NKX2-5, в предсердиях и проводящей системе сердца ограничена. При некоторых ВПС инактивируется связывание ДНК с белковым продуктом гена NKX2-5, что в 2 раза снижает физиологический уровень этого важнейшего транскрипционного фактора.
Двухстворчатый аортальный клапан
Недостаточное развитие трехстворчатого аортального клапана считают одним из наиболее распространенных ВПС, поражающим 2% живорожденных детей. Обычно это нарушение носит спорадический характер, однако наличие семейных форм заболевания свидетельствует об аутосомно-доминантной передаче.
К развитию двухстворчатого аортального клапана могут привести мутации гена NOTCH1, кодирующего регулятор транскрипции; у больных имеется склонность к прогрессирующей постнаталыюй калышфикации таких клапанов. Среди клинических проявлений, характерных для носителей мутации, отмечают стеноз аорты и недостаточность аортального клапана. Реже мутации гена NOTCH1 вызывают гипоплазию ЛЖ или тетраду Fallot.
Ген NOTCH1 является членом семейства генов, кодирующих трансмембранные рецепторы, которые выполняют различные функции в контроле дифференциации клетки и ее метаболизма. При активации кодируемого геном NOTCH1 рецептора происходит стимуляция протеолитического расщепления и высвобождение доменов, которые перемещаются к ядру клетки и участвуют в активации процессов транскрипции.
У человека мутации кодируемых геном NOTCH рецепторов, лигандов и ферментов вызывают различные заболевания с широким спектром стенотипических проявлений. Мутации этого гена, идентифицированные при наследственной форме развития двухстворчатого аортального клапана, инактивируют рецептор. И хотя механизм, согласно которому инактивация рецептора приводит к развитию аномального клапана, неизвестен, позднее отложение кальция в измененном по форме клапане может быть вызвано недостаточным ингибировапием гена RUNX2, кодирующего регулятор транскрипции активности остеобластов.