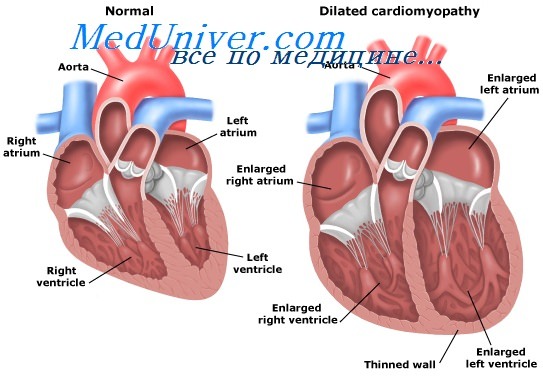
Генетические основы ДКМП. Механизмы развития идиопатический дилатационной кардиомиопатии
Мутация в бета-цепи MHС, приводящая к замене Ser-532Рrо, локализована в актинсвязывающем домене белка, который участвует в инициации сокращения. Изучение этой мутации с помощью биофизических методов позволило выявить снижение скорости движения актина вдоль мутантного миозина, снижение АТФазной активности и замедление скорости укорочения миоцита.
Эти начальные нарушения моторной функции мутантной формы миозина у мышей являются проявлениями сократительной дисфункции в виде морфологических и гистопатологических изменений при ДКМП. Следствием мутаций, затрагивающих белки тонких филаментов и связанных с ДКМП, является также снижение силы сокращения. При изучении мутаций в тропонине Т, тропонине С и альфа-тропомиозипе in vitro было обнаружено снижение активации АТФазы наряду со снижением генерации усилия. Снижение сократимости в результате мутаций саркомерных белков, вызывающих ДКМП, принципиально противоположно эффекту при мутациях, связанных с ГКМП.
Многие измутаций человека, связанных с ДКМП, возникают в специфических доменах гигантского мышечного белка титина, который взаимодействует со структурными элементами саркомера. Ассоциированные ДКМП мутации локализуются в кардиоснецифичном N2B-домене белка, который взаимодействуете I-полосами, растяжимыми участками тонких филаментов, обрамляющих Z-диски, и могут вызывать нарушения передачи усилия мышечного сокращения. В зоне Z-дисков титин взаимодействует с альфа-актинином, телетонином (кодируемым геном TCAP), мышечным LIM-бслком (кодируемым геном CLP) и специфическим белком cipher ZASP (кодируемым геном LBD3).
Имеются данные о том, что мутации, затрагивающие белки, взаимодействующие с I-дисками, вызывают у человека ДКМП. У некоторых носителей мутации из пораженных семей заболевание полностью не проявлялось. Фенотипические проявления, ассоциированные с указанными мутациями, ограничиваются миокардом, однако повышение уровня фермента в сыворотке крови может свидетельствовать о субклиническом течении миопатии скелетных мышц, что согласуется с общей ролью всех белков I-дисков во всех поперечнополо сатых мышцах. Мутации, затрагивающие белки Z-дисков, могут либо препятствовать их взаимодействию с титаном, либо дестабилизировать комплекс титин-Z-диск. В результате нарушаются нормальные детекция и модуляция механического напряжении, что приводит к развитию ДКМП.
Чтобы узнать, могут ли нарушения в передаче усилия между соседними миоцитами служить причиной развития ДКМП, изучали метавинкулин, изоформу белка винкулина, кодируемого геном VCL, картированным на хромосоме 10q22. Двемутации, обнаруженные у больных ДКМП, были найдены у непораженных родственников, что позволило предположить неполную пенентрантность этого гена. Иными словами, мутации тепа VCL редко становятся причиной ДКМП.
Доминантно наследуемые мутации белка фосфоламбан (продукта PLN-гена, локализованного на хромосоме 6q22) вызывают ДКМП за счет нарушений в регуляция уровня Са2+. У пораженных пациентов рано развиваются ДКМП и СН и возникает потребность в пересадке сердца уже в среднем возрасте. Фосфоламбан — это обратимый ингибитор Са2+-АТФазы сердечного саркоэндоплазматического ретикулума, который регулирует базальную сократимость миокарда. Ингибирование фосфоламбаном происходит в ответ на фосфорилирование путем стимуляции сердечных бета-адренорецепторов.
Доминантно наследуемые мутации гена PLN, вызывающие развитие ДКМП, могут предотвратить фосфорилирование или вызвать суперингибирование; любое изменение приведет к хронической задержке поглощении Са2+ после сокращения саркомера.
Ассоциированные с ДКМП человека мутации в регуляторной субъединице SUK2A (кодируемой геном ABCC9) АТФ-чувствительных калиевых каналов (КАГФ) Moiyr снизить способность сердца реагировать па стресс. КАТФ-каналы представляют- собой мультимерные белки, состоящие из самой канальной норы, обладающей способностью выпрямлять направленный внутрь ноток калия (Kir6.2), регуляторной субъединицы SUR2A и АТФ-связывающсго кассетного белка, удерживающего АТФазу. В сердечной мышце эти каналы участвуют в распознавании метаболических сигналов в ходе адаптации к стрессу. Обнаруженные у человека мутации делают КАТФ-каиалы нечувствительными к АДФ-индуцированным конфирмационным изменениям, которые модулируют открывание и закрывание канала.
Поскольку сердце, полученное от мыши с отсутствием гена КАТФ, подвержено перегрузке кальцием, некоторые патофизиологические особенности человеческих мутаций КАТФ (ДКМП и аритмии) могут отражать увеличение уровни кальция, индуцированное сокращением (напряжением) миоцитов.