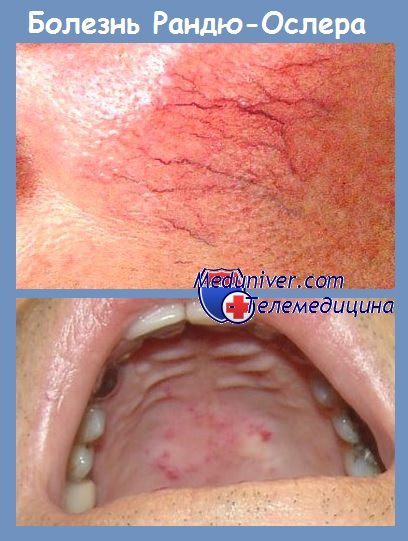
Врожденная геморрагическая телеангиэктазия (ВГТ) — это аутосомно-доминантное заболевание, ранее носившее название «болезнь Osler-Rendu-Weber». Его распространенность гораздо шире, чем полагают, и составляет 2-3 случая на 10 тыс. населения. Вследствие значительной межсемейной и внутрисемейной вариабельности это заболевание может оставаться нераспознанным в течение многих лет, даже несмотря на наличие у пациентов слабо выраженных симптомов.
Телеангиэктазии диаметром 0,5-3,0 мм, расположенные на коже и слизистых оболочках, обычно локализуются на языке, губах, туловище и кончиках пальцев. Артериовенозные фистулы мелкого или срелнего калибра обнаруживают в носу, тле они служат причиной рецидивирующих носовых кровотечений; в ЖКТ они также вызывают рецидивирующие кровотечения, сопровождающиеся скрытой анемией; в легких фистулы индуцируют гипоксемию, кровохарканье, полицитемию, способствуют утолщению концевых фаланг пальцев по типу «барабанные палочки», парадоксальной эмболии за счет шунта справа налево и гипердинамической циркуляции.
Реже сосудистые структурные изменения обнаруживают в головном мозге, печени и почках. У олного больного была найдена диффузная эктазия КЛ, у другого — геморрагический перикардит с тампонадой сердца.
Больным врожденной геморрагической телеангиэктазией и их близким родственникам показан скрининг для выявления артериовенозных мальформаций (АВМ) в легких с использованием контрастной ЭхоКГ. Появление «пузырьков» в левом предсердии в течение 4-10 сердечных циклов является характерным признаком наличия легочного шунта.
При положительном результате контрастной ЭхоКГ необходимо выполнить КТ-ангиографию высокого разрешения, чтобы документировать количество, размеры и локализацию АВМ. При любой мальформации, питающей артерии диаметром > 1 мм, следует предпринять попытку терапевтической эмболизации с тем, чтобы остановить снижение содержания кислорода в крови (гипоксемию) и предотвратить системную эмболизацию, особенно попадание эмболов в сосуды головного мозга.
В нескольких случаях удавалось уменьшить носовое и желудочно-кишечное кровотечения с помощью антифибринолитической терапии даназолом или аминокапроновой кислотой. Необходимость проведения контролируемых исследований по применению различных стратегий длительной терапии с учетом клинических и генетических переменных абсолютно очевидна. Мальформации сосудов головного мозга при ВГТ встречаются в < 25% случаев; обычно их обнаруживают в детском возрасте, и с годами они не увеличиваются в размерах.
Иными словами, МРТ необходимо назначать при малейшем подозрении на ВГТ и не повторять, если не обнаружено никаких сосудистых изменений или абсцессов, вызываемых паралоксалыюй эмболизацией септических сгустков, и не появляются новые неврологические симптомы или признаки. Встречаются сосудистые мальформации спинного мозга, но они довольно редки.
Можно предположить существование по меньшей мере 6 генов-кандидатов, обусловливающих BIT; 5 из них были картированы, а 3 удалось идентифицировать. Это тепы, кодирующие белки сигнальных путей — TGF-бета и белок морфогенеза костной ткани BMP (hone morphogenetic protein). Один локус, ENG, кодирует TGF-бета-связывающий белок, называемый эндоглином; в различных семьях обнаруживают различные мутации, связанные с ВГТ.
Другой локус, ALK1, кодирует активин-рецептор-подобную кииазу, которая относится к семейству сериновых-треониновых киназ, экспрессирующихся в эндотелиальных клетках. Можно предположить, что дефекты этого гена могут влиять на развитие и восстановление сосудов. У больных с BIT были обнаружены сотни мутаций в генах ENG и ALK, но распространены они только у 65-75% больных. У больных с аутосомно-доминантным ювенильным полипозом ЖКТ, вызванным мутациями гена SMAD4, обнаруживают много общих признаков с ВГТ. Таким образом, любой пациент с ювенильным полипозом должен быть обследован на наличие этого заболевания.
Четвертый локус ВГТ был картирован на коротком плече 7-й хромосомы, но идентифицировать ген не удалось; вероятно, будут обнаружены и другие, более редкие локусы. Однако уже сейчас проведение доклинической и пренатальной диагностики путем выявления мутаций или анализа сцепления генов доступно широкому числу пациентов с потенциально жизнеугрожающим заболеванием.
Наиболее распространенной причиной артериовенозных мальформаций, наследуемых по законам Менделя, являются различные формы BIT. Одновременно изолированные артериовенозные структурные нарушения, особенно в головном мозге, можно встретить довольно часто; существуют и другие примеры наследственной предрасположенности, например синдром Parkes-Weber.