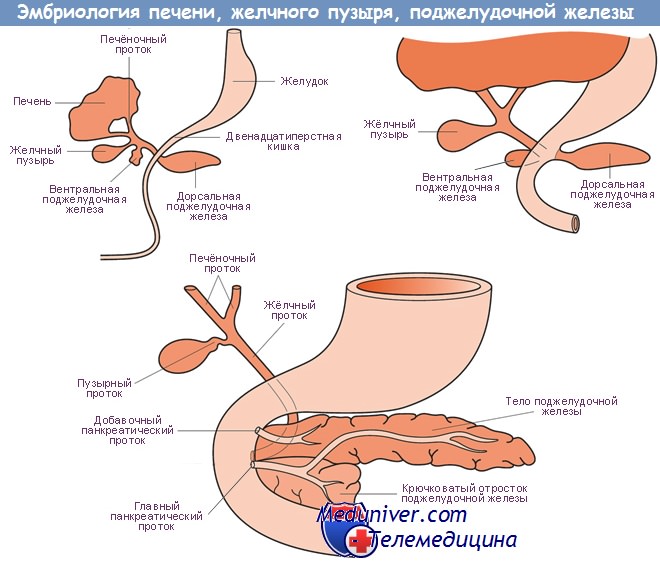
У млекопитающих зрелая поджелудочная железа образуется в результате слияния дорсального и вентрального зачатков, что происходит одновременно с поворотом средней кишки. В процессе слияния двух зачатков образуется анастомоз между их протоками, причем вентральный проток в норме служит для выведения панкреатического секрета из обеих частей железы. Примерно у 9% людей протоки не анастомозируют полностью.
При этом сохраняются две отдельные протоковые системы, что способствует возникновению острой и хронической недостаточности поджелудочной железы. Такое сохранение разделения поджелудочной железы наблюдали в эксперименте у гетерозиготных мышей, имеющих гены Ibb и Sbb. Эти исследования позволили предположить, что сигнальный путь hedgehog может играть определенную роль в возникновении указанной патологии.
Мутации этих генов также вызывают избыточный рост вентрального зачатка поджелудочной железы и могут привести к образованию кольцевидной поджелудочной железы — врожденного дефекта, который часто является причиной дуоденальной непроходимости.
Описана полная и частичная агенезия поджелудочной железы в результате мутаций, определяющих последовательность гена IPF1, и снижения периода полураспада фактора-промотора инсулина. Агенезия дорсальной части поджелудочной железы была выявлена у мышей, имеющих мутацию Raldh2, обусловленную дефицитом ретиноевой кислоты. Эти патологические состояния часто сопровождаются диабетом и могут сочетаться с сакральной агенезией.
Полная панкреатическая агенезия сочетается с экзокринной недостаточностью поджелудочной железы и диабетом и обычно является фатальным заболеванием.
Были описаны редкие случаи изолированного дефицита энтерокиназы (которая необходима для превращения трипсиногена в трипсин), трипсиногена, липазы и колипазы. Обычно у пациентов с указанной патологией заболевание протекает тяжело и трудно поддается лечению. Симптомы мальабсорбции проявляются сразу после рождения.
Синдромы, включающие аномалии развития поджелудочной железы
Ряд синдромов сочетается с недостаточностью поджелудочной железы или аномалиями ее развития и функции, например фиброзом. Однако симптомы панкреатической недостаточности редко присутствуют в периоде новорожденности.
Синдром Швахмана-Дайемонда — аутосомно-рецессивное заболевание, которое проявляется жировой дегенерацией поджелудочной железы, дисфункцией костного мозга, изменениями в печени, костной и других системах. Симптомы поджелудочной недостаточности в неонатальном периоде обычно не проявляются. Ген SDS идентифицирован и клонирован. Известно, что он локализуется на хромосоме 7.
Синдром Иогансона—Близзарда — редкое аутосомно-рецессивное заболевание, при котором отмечаются экзокринная панкреатическая недостаточность вследствие отсутствия панкреатических ацинусов при интактных протоках, аплазия или гипоплазия крыльев носа и дефект кожи на голове. Синдром Иогансона-Близзарда обусловлен мутацией гена UBRI, локализованного на хромосоме 15q. Признаками также могут быть глухота, гипотироидизм, отсутствие зубов, задержка развития, сердечные и аноректальные аномалии.
Эндокринопатия может проявляться гипопитуатаризмом, дефицитом гормона роста, сахарным диабетом. Если синдром устанавливается с рождения, то присутствует большое количество врожденных аномалий, а панкреатическая недостаточность может быть причиной диареи и мальабсорбции.
Синдром Жена (асфиксическая дистрофия грудной клетки) представляет собой сочетание дистрофии грудной клетки, карликовости с укорочением конечностей и кистозной дисплазии почек. В некоторых случаях выявляли экзокринную панкреатическую недостаточность. Гистологическая картина у некоторых пациентов напоминала таковую при синдроме Швахмана-Дайемонда. У пациентов с синдромом Жена необходимо оценить сохранность функции поджелудочной железы, а также исключить синдром Швахмана-Дайемонда.
Синдром Пирсона — это митохондриальное нарушение, характеризующееся дисфункцией костного мозга и нарушением экзокринной функции поджелудочной железы. Как и при иных видах митохондриальной патологии, могут быть поражены другие органы и системы. Обычно в неонатальном периоде симптомы панкреатической недостаточности не отмечаются.
При некоторых синдромах (например, синдроме Ивемарка, трисомии 9, синдроме Меккеля, хондродисплазии Жена и Сальдино-Нунана, а также глютаровой ацидемии типа II) встречается сочетание аномалий почек, печени и поджелудочной железы. Bernstein и соавт. сделали вывод, что после исключения определенных синдромов случаи почечно-печеночно-панкреатической дисплазии не обязательно составляют гомогенную группу. Несмотря на наличие дисфункции поджелудочной железы, она обычно не является основным синдромом.
Гиперинсулинемическую гипогликемию (старое название — незидиобластоз), причиной которой является распространенная или локальная гиперфункция b-клеток, часто лечат путем тотальной резекции поджелудочной железы или панкреатэктомии 95% объема органа. Такое лечение предполагает развитие в дальнейшем экзокринной панкреатической недостаточности, поэтому пациенты, перенесшие подобную операцию, нуждаются в оценке экзокринной функции поджелудочной железы.
Учебное видео по развитию желудочно-кишечного тракта (эмбриогенезу)