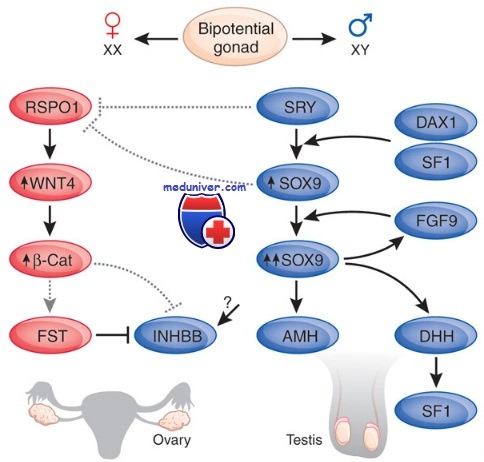
Мутации и дупликации генов DAX1, SOX9. Несоответствие пола генотипу XY и кампомелическая дисплазия
DAX1 — Х-сцепленный ген, экспрессирующийся развивающимся эмбрионом. Специфические органы его экспрессии — гипоталамус, гипофиз, надпочечники и гонады. Нарушения экспрессии DAX1 проявляются в несоответствии пола и врожденной гипоплазии надпочечников. DAX1 — ядерный рецепторный белок, получивший свое название от англоязычных аббревиатур, обозначающих:
• D (dosage-sensitive sex reversal) — дозозависимое несоответствие пола;
• A (adrenal hypoplasia congenita) — врожденная гипоплазия надпочечников;
• X — критический регион Х-хромосомы;
• 1 — ген 1.
По-видимому, DAX1 принадлежит к группе генов-антагонистов развития по мужскому типу. Мутации или делеции гена DAX1 вызывают врожденную гипоплазию надпочечников. Дупликации этого гена приводят к развитию несоответствия пола у фенотипических женщин с генотипом XY. Мутации гена DAX1 ответственны за возникновение врожденной гипоплазии надпочечников с надпочечниковой недостаточностью; патология развития гипофиза приводит к гипогонадотропному гипогонадизму.
Несоответствие пола генотипу XY
Дупликация региона DAX1 может приводить к несоответствию пола генотипу XY и блоку развития яичек (дисгенезия гонад). Гонада, не содержащая сперматозоидов или ооцитов, называется полосковидной гонадой. Частичная дисгенезия гонад может развиваться при условии сохранения части функционально активных тканей яичка. Наружные гениталии при этом могут быть бисексуальными. Фенотипические мужчины с несоответствием пола характеризуются отсутствием мюллерова ингибирующего фактора и персистенцией мюллеровых протоков. Этим они отличаются от фено-типических женщин с генотипом XY, нечувствительных к андрогену, у которых отсутствуют остаточные мюллеровы структуры.
У больных с чистой дисгенезией гонад с генотипом XY повышен риск гонадобластомы, встречающейся у них с частотой 10-30%. Большинство случаев чистой дисгенезии гонад обусловлено мутациями гена SRY. Обследование таких пациентов должно включать анализ кариотипа на предмет делеций Х-хромосомы, анализ гена SRY на предмет мутаций и молекулярное или FISH-тестирование на предмет дупликаций гена DAX1. В комплекс лечения следует включить заместительную терапию эстрогенами задержки полового созревания.
Кампомелическая дисплазия и мутации гена SОX9
Ген SOX9 был описан в 1994 г. как регулятор половой дифференцировки по мужскому типу, участвующий также в процессах развития скелета. Ген SOX9 расположен на длинном плече хромосомы 17 и в некоторой степени гомологичен гену SRY, отсюда его название — SOX, т.е. «бокс, связанный с полом».
Он выступает в качестве фактора транскрипции и экспрессируется у человека в яичке, почках, хондроцитах, печени и головном мозге. SOX9 играет важнейшую роль в дозозависимой половой дифференцировке по мужскому типу, что было показано при гаплонедостаточности SOX9, приводящей либо к несоответствию пола, либо к амбивалентной половой дифференцировке. Повышение дозы SOX9 за счет дупликации вызывает аутосомное несоответствие пола у лиц с генотипом XX. Кроме того, SOX9 участвует в активации мюллерова ингибирующего фактора, а также играет роль в развитии костной и хрящевой ткани, вероятно, являясь регулятором экспрессии COL2A1, как показано на больных с аномалиями скелета при кампомелической дисплазии.
Кампомелическая дисплазия — спорадическое заболевание, характеризующееся низким ростом, короткими конечностями, изогнутыми нижними конечностями, нарушениями строения лица, расщеплением нёба и зачастую врожденными пороками сердца. Нередки летальные исходы, связанные с мутациями гена SOX9. Считают, что заболевание наследуется по аутосомно-доминантному типу, при этом для него высока частота мутаций de novo за счет высокой летальности. Как и для многих других синдромов с предполагаемым гонадным мозаицизмом, риск повторного возникновения заболевания для пары составляет около 3%.