Миотубулярная миопатия у детей. Диагностика и лечение
Термин миотубулярная миопатия предполагает остановку созревания фетальных мышц на миотубулярной стадии развития, которая соответствует 8-15 нед. внутриутробного периода. Это основано на морфологическом изменении мышечных волокон: в центре цитоплазмы расположен ряд ядер, вокруг которого сократимые миофибриллы формируют цилиндр. Многие ученые подвергают сомнению эту интерпретацию и применяют по отношению к этому виду миопатий более нейтральный термин центронуклеарная миопатия. Однако этот термин неспецифичен, так как внутренние ядра наблюдаются при многих видах миопатий, не связанных между собой.
Хотя в патогенезе этого состояния иногда предполагается неврогенный компонент, характерны нормальное число и морфология мотонейронов спинного мозга и, как правило, нормальная ультраструктура периферических нервов и скорость проведения по нервам. Стойкое повышение концентрации фетального виментина и десмина отмечается в мышечных волокнах младенцев с миотубулярной миопатией, хотя не выявляется в культурах миоцитов пациентов. Эти белки промежуточных филаментов служат элементами цитоскелета миотубул плода, присоединяя ядра и митохондрии к сарколемме и сохраняя их центральное расположение.
При изменении внутриклеточной организации в процессе созревания мышц ядра перемещаются к периферии, а митохондрии перераспределяются между миофибриллами. В то же время количество виментина и десмина уменьшается. К моменту рождения виментин полностью исчезает, а десмин остается в следовых количествах. Сохранение фетального виментина и десмина в мышечных волокнах может служить одним из механизмов остановки созревания мышечной ткани.
Вторичный миастеноподобный дефект нервно-мышечной передачи также возникает у некоторых младенцев с миотубулярной миопатией. Миоциты пациентов, культивируемые in vitro совместно с нервными волокнами, иннервируются и созревают нормально, и патологические изменения, найденные in vivo, отсутствуют.
Клинические проявления миотубулярной миопатии у детей
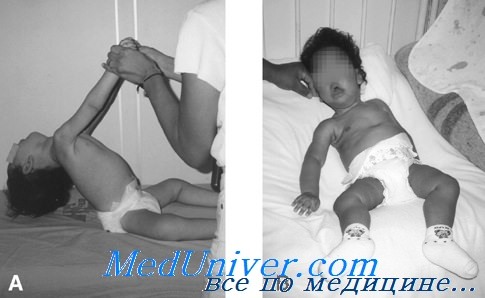
На поздних сроках гестации может отмечаться снижение двигательной активности плода. Частым осложнением во время беременности служит многоводие, которое связано со слабостью ротоглоточной мускулатуры плода и неспособностью заглатывать амниотическую жидкость. При рождении отмечаются снижение мышечной массы, включая аксиальную мускулатуру, мышцы плечевого и тазового пояса и дистальные группы мышц, генерализованная мышечная гипотония и диффузная слабость. Дыхательные движения могут быть неэффективными, что требует ИВЛ.
Может потребоваться зондовое питание в связи со слабостью мышц, осуществляющих сосание и глотательные движения. Часто выявляется крипторхизм. Возможна слабость мимической мускулатуры, однако для этих младенцев нехарактерны черты лица, свойственные миотонической дистрофии. В отдельных случаях наблюдается офтальмоплегия. Может выявляться высокое готическое нёбо. Язык тонкий (атрофия мышц языка), однако фасцикуляции в языке отсутствуют. Сухожильные рефлексы снижены или отсутствуют. Для миотубулярной миопатий кардиомиопатия нехарактерна; зрелым волокнам сердечной мышцы в норме свойственно центральное расположение ядер. Врожденные аномалии центральной нервной системы или других органов нехарактерны.
У детей более старшего возраста и взрослых может развиваться центронуклеарная миопатия с вариабельной мышечной слабостью. Связь этого заболевания с тяжелой формой миотубулярной миопатий новорожденных в настоящее время не до конца ясна.
Лабораторные исследования при миотубулярной миопатии у детей. Активность креатинфосфокиназы (КФК) в крови в пределах нормы. ЭМГ не выявляет признаков денервации; результаты чаще в пределах нормы, или с раннего младенческого возраста выявляются минимальные неспецифические признаки. Скорость проведения возбуждения по нервам может быть снижена, однако, как правило, в пределах нормы. ЭКГ без патологии. Рентгенография грудной клетки не обнаруживает признаков кардиомегалии; ребра могут быть тонкими.
Диагностика миотубулярной миопатии у детей
Биопсия мышц имеет диагностическое значение уже при рождении, даже у недоношенных новорожденных. Более 90 % мышечных волокон уменьшено в размерах и имеют крупные везикулярные ядра, локализованные в центральной области и расположенные в один ряд. Пространства между ядрами заполнены саркоплазмой, содержащей митохондрии. Гистохимическое окрашивание для выявления активности окислительных ферментов и гликогена демонстрирует центральное распределение, характерное для миотубул плода. В цилиндрах миофибрилл обнаруживается зрелая гистохимическая дифференцировка при окрашивании на АТФазу.
Соединительная ткань в составе мышц, мышечных веретен, кровеносных сосудов, внутримышечных нервных волокон, концевых пластинок двигательных нервов имеет зрелую структуру. Ультраструктурная картина при неонатальной миотубулярной миопатии, за исключением специфических диагностических признаков, также характерна для зрелой мышечной ткани. Высокая иммунореактивность виментина и десмина бывает в мышечных волокнах при миотубулярной миопатии, в то время как в мышцах здорового доношенного новорожденного активность этих ферментов отсутствует.
Разработаны методы определения молекулярно-генетических маркеров миотубулярной миопатии в крови. Молекулярно-генетическое исследование проводится не только с целью подтверждения диагноза, но и для ранней диагностики в пренатальном периоде.
Генетика миотубулярной миопатии у детей. Наиболее распространен Х-сцепленный рецессивный тип наследования; большинство пациентов — мальчики. У матерей младенцев с миотубулярной миопатией клинические проявления заболевания отсутствуют. Однако при мышечной биопсии выявляются минимально выраженные изменения. Зарегистрированы случаи заболевания с аутосомно-доминантным и аутосомно-рецессивным типом наследования, однако их распространенность низка. Ген картирован на Х-хромосоме в локусе Xq28, отличном от локуса Хр21 при мышечной дистрофии Дюшенна и Беккера.
Идентифицирована делеция в гене МТМ1, ответственная за развитие заболевания. Этот ген кодирует белок миотубулярин с доменом пируваттирозинфосфатазы. Хотя поражение локализуется только в одном гене, выявлено пять различных точечных мутаций гена МТМ1 (из 242 известных мутаций генов), которые определяют развитие заболевания только в 27 % случаев; мутации многих различных аллелей могут вызывать сходные клинические проявления заболевания.
Лечение миотубулярной миопатии у детей. Поддерживающая и паллиативная терапия.
Прогноз миотубулярной миопатии у детей. Около 75 % новорожденных с тяжелой формой заболевания умирает в первые несколько недель или месяцев жизни. У выживших детей заболевание не прогрессирует, однако имеется выраженное нарушение двигательной функции; дети редко овладевают навыком ходьбы, сохраняется тяжелая гипотония.