Для MERRF-синдрома характерна прогрессирующая миоклонус-эпилепсия, митохондриальная миопатия, мозжечковая атаксия, дизартрия и нистагм. Дебют бывает в детском возрасте или у взрослых; возможно медленно прогрессирующее течение или быстрое развитие заболевания. Другие симптомы заболевания включают деменцию, нейросенсорную тугоухость, атрофию зрительных нервов, периферическую невропатию и спастичность.
Поскольку у некоторых пациентов определяются нарушение глубокой чувствительности и деформация стоп (pes cavus), в этих случаях может ошибочно диагностироваться атаксия Фридрейха. У пациентов с MELAS- и MERRF-синдромом бывают случаи заболевания в семейном анамнезе и низкий рост. Характерен материнский тип наследования.
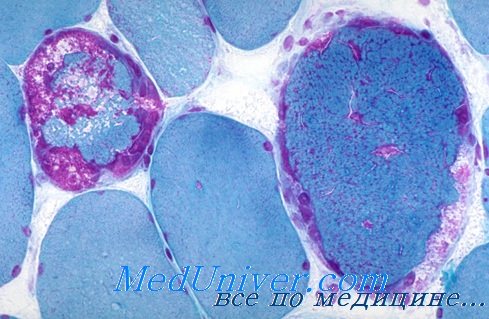
У пациентов с MERRF-синдромом определяются повышение уровня лактата в сыворотке крови, феномен рваных красных волокон при мышечной биопсии, при морфологическом исследовании — выраженная гибель нейронов и глиоз, особенно в области зубчатого ядра и нижней оливы, признаки гибели клеток Пуркинье и нейронов красного ядра. Отмечаются побледнение задних канатиков спинного мозга и дегенерация тонкого и клиновидного ядер.
Мышечная биопсия обнаруживает вариабельный дефицит комплекса III, комплексов II и IV, комплексов I и IV и изолированный дефицит комплекса IV. Более чем в 80 % случаев заболевание вызвано гетероплазмической точечной мутацией в позиции 8344 гена лизиновой тРНК (нуклеотидная замена G—>А). У некоторых пациентов выявляется мутация в нуклеотиде 8356 гена лизиновой тРНК (нуклеотидная замена Т—>С).
Специфическое лечение отсутствует, хотя описан хороший терапевтический эффект коэнзима Q10 у матери и дочери с MERRF-мутацией.
Синдром LHON дебютирует обычно у пациентов в возрасте 18—30 лет с острой или подострой потери зрения, вызванной тяжелой билатеральной атрофией зрительных нервов, хотя имеются сообщения о начале заболевания у детей в возрасте 5 лет. Практически 85 % пациентов составляют мужчины молодого возраста. Это позволяет предположить Х-сцепленный фактор, модулирующий экспрессию точечной мутации мтДНК.
Классические симптомы при офтальмологическом обследовании включают циркумпапиллярную (вокруг диска зрительного нерва) телеангиэктатическую микроангиопатию и псевдоотек диска зрительного нерва. Непостоянные признаки заболевания включают мозжечковую атаксию, гиперрефлексию, положительный симптом Бабинского, психические нарушения, периферическую невропатию и нарушение сердечной проводимости (синдром перевозбуждения желудочков). В некоторых случаях отмечается распространенное поражение белого вещества, характерное для рассеянного склероза. Синдром LHON отличается отсутствием лактат-ацидоза и рваных красных волокон.
При этом заболевании описано более 11 точечных мутаций мтДНК, включая гомоплазмическую мутацию в нуклеотиде 11 778 (нуклеотидная замена G—>А) в структуре ND4 субъединицы гена комплекса I дыхательной цепи. Мутация представляет собой замену высококонсервативного аргинина на гистидин в позиции 340-й аминокислоты и служит причиной синдрома LHON примерно в 50-70 % случаев в Европе и более чем в 90 % случаев в Японии. Описаны родословные пациентов с синдромом LHON, в основе которого лежат другие точечные мутации; в этих случаях характерные проявления синдрома сочетались с комплексом неврологических нарушений, клиническая картина заболевания напоминала синдром MELAS и младенческий билатеральный некроз полосатого тела.
Мутация в субъединице 6 митохондриальной АТФазы (NARP) заболевание с материнским типом наследования проявляется в виде фенотипической картины синдрома Ли или в виде совокупности симптомов: задержка развития, пигментный ретинит, деменция, судороги, атаксия, слабость мышц проксимальных отделов конечностей и сенсорная невропатия (NARP-синдром). Заболевание вызвано точечной мутацией в нуклеотиде 8993 гена субъединицы 6 АТФазы. Выявляется близкая корреляция тяжести клинических проявлений заболевания и долей мутантной мтДНК в лейкоцитах.