Гликогенозы у детей: болезни Гирке, Форбса-Кори, Андерсена, Мак-Ардла, Таруи
Гликогеноз I типа (болезнь Гирке) — не истинная миопатия, а заболевание, обусловленное дефицитом печеночного фермента глюкозо-6-фосфатазы, который в норме не присутствует в мышце, тем не менее у детей с этим заболеванием выявляется гипотония и умеренно выраженная мышечная слабость неизвестной этиологии. Гликогеноз II типа (болезнь Помпе) — аутосомно-рецессивно унаследованный дефицит гликолитического лизосомного фермента кислой мальтазы. Из 12 известных типов гликогенозов только II тип обусловлен дефектом лизосомного фермента. Аномальный ген картирован в локусе 17q23. Описано две формы заболевания.
Младенческая форма характеризуется тяжелой генерализованной миопатией и кардиомиопатией. У пациентов выявляется кардиомегалия, гепатомегалия, диффузная гипотония и мышечная слабость. Активность КФК в крови значительно повышена. Мышечная биопсия выявляет вакуолярную миопатию в сочетании с нарушением активности ферментов лизосом, таких как кислая и щелочная фосфатазы. Смерть обычно наступает в младенческом или раннем детском возрасте.
Поздняя детская или взрослая форма представлена миопатией с более легким течением, без увеличения сердца и печени. Клинические проявления могут отсутствовать до позднего детского или раннего зрелого возраста, однако возможно появление признаков мышечной слабости (обусловленной миопатией) и гипотонии даже в раннем младенческом возрасте. Активность КФК в крови значительно повышена, результаты мышечной биопсии имеют диагностическое значение даже на пресимптомной стадии заболевания.
Диагноз гликогеноза II типа подтверждается при количественном анализе активности кислой мальтазы при биопсии мышц или печени. При редком варианте дефицита кислой мальтазы с легким течением ее активность при биопсии мышц может находиться на нижней границе нормы с периодическим снижением до субнормального уровня, при этом результаты мышечной биопсии напоминают гликогеноз II типа, но изменения выражены более умеренно.
Другая форма — болезнь Данона — характеризуется Х-сцепленным рецессивным типом наследования, аномальный ген картирован в локусе Xql4. В основе заболевания лежит первичный дефицит протеина-2 мембран лизосом (LAMP2), который приводит к развитию гипертрофической кардиомиопатии, миопатии с поражением мышц проксимальных отделов конечностей и умственной отсталости.
Гликогеноз III типа (болезнь Форбса-Кори) обусловлен дефицитом фермента, расщепляющего гликоген (амило-1,6-глюкозидаза). Это наиболее распространенный гликогеноз с наименее тяжелыми клиническими проявлениями по сравнению с другими типами гликогенозов. В младенческом возрасте часто встречаются такие симптомы, как гипотония, мышечная слабость, гепатомегалия, гипогликемия при исследовании крови натощак, однако эти симптомы часто спонтанно исчезают и в детском возрасте, а также у взрослых клинические проявления могут отсутствовать.
В других случаях отмечается медленное прогрессирование атрофии мышц дистальных отделов конечностей, цирроза печени и сердечной недостаточности. При мышечной биопсии обнаруживаются минимально выраженные миопатические изменения, включающие вакуолизацию мышечных волокон.
Гликогеноз IV типа (болезнь Андерсена) обусловлен дефицитом фермента, участвующего в синтезе гликогена, приводящего к синтезу аномальных молекул гликогена — амилопектина — в печени, ретикулоэндотелиальных клетках, скелетной мускулатуре и сердечной мышце. Гипотония, генерализованная мышечная слабость, атрофия мышц и контрактуры — характерные признаки миопатического процесса. Большинство пациентов умирает до 4-летнего возраста в связи с развитием печеночной или сердечной недостаточности. Описаны отдельные случаи заболевания у детей без признаков нервно-мышечного заболевания.
Гликогеноз V типа (болезнь Мак-Ардла) обусловлен дефицитом мышечной фосфорилазы, наследуемым по аутосомно-рецессивному типу, аномальный ген картирован в локусе 1lql3. Основным клиническим проявлением заболевания служит непереносимость физической нагрузки, которая вызывает болезненный мышечный спазм (крампи), мышечную слабость и миоглобинурию; однако между приступами мышечная сила не снижена. Активность КФК в крови повышена только во время физической нагрузки. Характерным клиническим признаком служит отсутствие наблюдаемого в норме повышения уровня лактата в крови во время физической нагрузки, приводящей к ишемии.
Это обусловлено невозможностью превращения пирувата в лактат при анаэробных состояниях in vivo. Дефицит миофосфорилазы можно обнаружить с помощью гистохимических и биохимических методов в мышечном биоптате.
Редкая неонаталъная форма дефицита миофосфорилазы вызывает бульбарные расстройства в раннем младенческом возрасте, которые могут быть настолько выражены, что приводят к летальному исходу в периоде новорожденное™. В других случаях возможно медленное прогрессирование мышечной слабости, напоминающее мышечную дистрофию.
Отдаленный прогноз благоприятный. Пациенты должны научиться контролировать свой уровень физической нагрузки; тяжелой инвалидизации вследствие хронической миопатии или поражения сердца не отмечается.
Гликогеноз VII (болезнь Таруи) представляет собой дефицит мышечной фосфофруктокиназы. Хотя это заболевание встречается реже, чем гликогеноз V типа, оба заболевания характеризуются непереносимостью физической нагрузки, похожим клиническим течением и невозможностью превращения пирувата в лактат. Биохимическое исследование мышечных биоптатов позволяет дифференцировать эти два типа гликогенозов. Заболевание наследуется по аутосомно-рецессивному типу, аномальный ген картирован в локусе lcenq32.
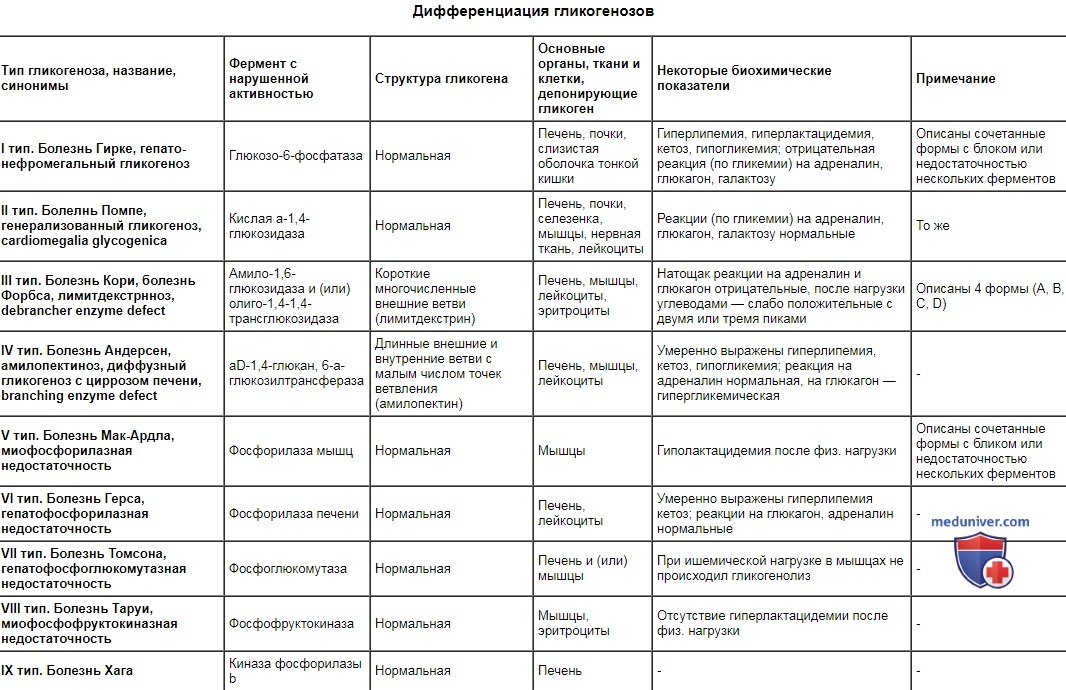
Дифференциация гликогенозов
Тип гликогеноза, название, синонимы
Фермент с нарушенной активностью
Структура гликогена
Основные органы, ткани и клетки, депонирующие гликоген
Некоторые биохимические показатели
Примечание
I тип. Болезнь Гирке, гепато-нефромегальный гликогеноз
Глюкозо-6-фосфатаза
Нормальная
Печень, почки, слизистая оболочка тонкой кишки
Гиперлипемия, гиперлактацидемия, кетоз, гипогликемия; отрицательная реакция (по гликемии) на адреналин, глюкагон, галактозу
Описаны сочетанные формы с блоком или недостаточностью нескольких ферментов
II тип. Болелнь Помпе, генерализованный гликогеноз, cardiomegalia glycogenica