Известна аутосомно-рецессивная форма синдрома множественных делеций мтДНК - так называемая митохондриальная нейрогастроинтестинальная энцефаломиопатия (MNGIE). Заболевание манифестирует в более молодом возрасте и проявляется тем же набором типичных для митохондриальных болезней неврологических симптомов в сочетании с выраженной дисфункцией желудочно-кишечного тракта (синдром кишечной нсевдообструкции с приступами повторной рвоты, диареей, потерей массы тела).
У больных MNGIE выявлено выраженное снижение активности фермента тимидин-фосфорилазы, обусловленное мутациями соответствующего гена на хромосоме 22ql3.32-qter [Nishino I. et al., 1999]. Таким образом, в основе болезни лежит генетически детерминированная патология метаболизма тими-дина, приводящая к нарушению репликации и/или поддержания молекулы мтДНК.
Синдром истощения мтДНК представляет собой состояние, при котором у больных имеется не качественный, а количественный дефект мтДНК - т.е. резкое снижение числа копий молекул мтДНК (рис. 68 В) [Moracs С. el al, 1991; DiMauro S., 1993]. Характерно, что истощение мтДНК наблюдается только в строго определенных тканях (например, только в мышцах, только в печени, в мышцах и почках и т.д.).
Клиническая картина зависит от вовлечения конкретных тканей и обычно включает в различных комбинациях миопатию (в том числе врожденную), судорожный синдром, печеночную и почечную недостаточноть, кардиомиопатию. Характерен лактатацидоз, феномен «рваных красных волокон», обнаруживается комбинированная недостаточность комплексов дыхательной цепи, содержащих мтДНК-кодируемые субъединицы (I, III-V).
Заболевание носит врожденный характер или манифестирует на 1 -2-м году жизни и имеет обычно фатальное течение. В большинстве описанных случаев синдрома истощения мтДНК зарегистрирован аутосомно-рецессивный тип наследования. Генетический дефект не установлен. Предполагается, что заболевание обусловлено повреждением ядерного гена, контролирующего репликацию мтДНК. Известна также относительно доброкачественная вторичная форма данного синдрома, при которой истощение мтДНК в клетках вызвано применением анти-ВИЧ препарата зидовудина.
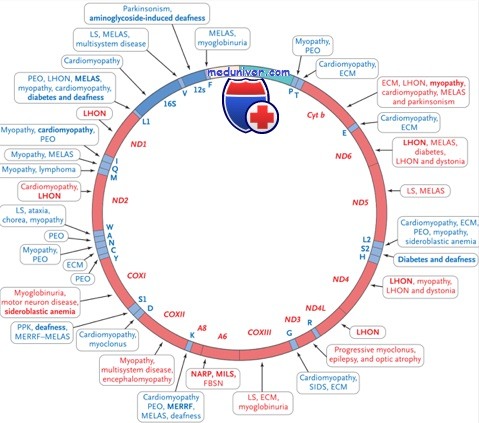
Виды патологии митохондриальной ДНК. Представлен участок отвечающий за развитие миоклонус-эпилепсии с рваными красными волокнами MERRF
В силу особенностей генетики митохондриальных энцефаломиопатий ДЫК-диагностика этих заболеваний имеет ряд принципиальных отличий от традиционных подходов, применимых для менделирующих заболеваний. Обобщение многолетнего опыта обследования боль,ных с митохондриалыюй патологией в ведущих исследовательских центрах мира позволило разработать четкий и последовательный диагностический алгоритм, обеспечивающий наибольшую эффективность диагностического поиска [Краснопольская К.Д., Захарова ЕЛО., 1998; Shoffner J., Wallace D., 1992; DiMauro S., 1993; Chinnery P. et al., 1999 (а)]. Данный алгоритм включает несколько основных этапов.
На первом этапе проводится детальный клинико-генеалогический и лабораторпо-инструментальный анализ, целью которого является накопление доказательств в пользу обоснованного предположения о митохондриальной природе изучаемого заболевания. Наиболее очевидно о митохондриальной болезни может свидетельствовать:
а) материнский тип наследования (с учетом всех полиморфных и даже субклинических проявлений у сибсов - детей больной матери);
б) своеобразный характер синдрома (наличие мультисистемной и мультиорганной патологии с вовлечением органов, различных по эмбриональному происхождению и функциям);
в) прогрессирующее течение, наличие метаболических кризов (последнее особенно актуально для митохондриальных синдромов раннего детского возраста);
г) повышение уровня лактата в крови и спинномозговой жидкости (в том числе на фоне пищевой и физической нагрузки);
д) аминоацидурия и органическая ацидурия.
Поиск мультиорганной патологии должен проводиться целенаправленно, с использованием необходимых параклинических методов, с целью выявления явной или скрытой кардиомиопатии, почечной тубулопатии, гепатоцеллюлярной дисфункции, диабета, недостаточности гормона роста, атрофии ворсин кишечника, изменений в мазке крови и пунктате костного мозга, миопатии, периферической, кохлеарной и зрительной невропатии, патологии сетчатки, петрификатов и очаговых изменений в веществе мозга.