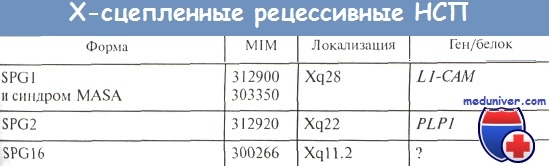
X-сцепленная спастическая параплегия. Генетические формы спастической параплегии
Вторая генетическая форма Х-сцепленной рецессивной спастической параплегии сцеплена с хромосомным участком Xq22 (локус SPG2) и обусловлена мутациями в гене протеолипидного белка (PLP) [Saugier-Veber P. et al., 1994]. Интересно отметить, что различные точковые мутации в данном гене приводят к манифестации как «чистой» спастической параплегии, так и осложненной ее формы (в сочетании с умственной отсталостью) [Cambi F. et al., 1996; Reid E., 1999]. Более того, мутации в гене PLP (чаще всего дупликации) являются молекулярной основой еще одного Х-сцепленного синдрома - болезни Пелицеуса-Мерцбахера, которая относится к группе лейкодистрофий и характеризуется врожденной гипотонией, нистагмом, задержкой психического развития, прогрессирующими пирамидными, мозжечковыми и дистоническими нарушениями [Saugier-Veber P. et al., 1994]. Продукт гена PLP - протеолипидный белок - играет ведущую роль в созревании олигодендроцитов и компактной укладке миелиновых слоев [Griffiths I. et al., 1995].
Предполагается, что дупликации и другие мутации, влияющие на функцию данного белка в отношении созревания олигодендроцитов, проявляются тяжелым фенотипом болезни Пелицеуса-Мерцбахера, тогда как мутации PLP, затрагивающие лишь процессы компактизации миелина, приводят к манифестации бонее легких аллельных заболеваний - «чистой» и осложненной форм спастической параплегии [Reid E., 1999]. Локусы аутосомно-рецессивных форм наследственной спастической параплегии картированы на хромосомах 8ql2—13 (SPG5), 16q24.3 (SPG7), 15ql3-15 (SPGll), 3q27-28 (SPG14) и 14q22-24 (SPG15). Ген формы SPG7, получивший название «параплегии», был недавно идентифицирован: он кодирует синтез АТФ-зависимой металлопротеазы, локализованной на внутренней мембране митохондрий и играющей роль в процессах протеолиза и организации/деградации белковых комплексов [Casari G. et al., 1998]. Как и спастин (продукт гена доминантной формы SPG4), белок параплегии относится к ААА-семейству клеточных АТФ-аз [Pearce D., 1999].
В случае мутаций в параплегине у больных с SPG7-формой заболевания выявляется системный дефект окислительного фосфорилирования, что может быть связано с нарушением сборки субъединиц комплексов дыхательной цепи митохондрий и приводить к дегенерации длинных аксонов центральных мотонейронов. Следовательно, 8РС7-форма аутосомно-рецессив-ной спастической параплегии является ядерно-кодируемым митохондриальным заболеванием. Для митохондриальных заболеваний характерен полиморфизм клинических проявлений, и SPG7 подтверждает данное правило, поскольку различные мутации параплегина могут манифестировать как в виде «чистых» форм спастической параплегии, так и в виде осложненных ее вариантов (в сочетании с атрофией зрительных нервов, атрофией коры мозжечка или больших полушарий мозга) [Reid E., 1999].
Такой же клинический полиморфизм характерен и для формы SPG11, ген которой локализован на 15-й хромосоме, но пока не идентифицирован: часть больных SPG11 имеют изолированную спастичность в ногах, тогда как в других случаях SPG11 спастический парапарез сочетается с агенезией мозолистого тела, умственной отсталостью и дизартрией [Martinez Murillo F. et al., 1999].
Для SPG5 характерна изолированная спастическая параплегия с ранним началом болезни [Hentati A. et al., 1994 (б)]. Форма SPG14 описана лишь в одной семье, в которой у всех больных спастичность манифестировала в сравнительно позднем возрасте (около 30 лет) и сочеталась с дистальной моторной невропатией и умственной отсталостью [Vazza G. et al., 2000]. Для SPG 15 (синдром Кьеллина) характерно сочетание спастичности с пигментной дегенерацией сетчатки, дистальными амиотрофиями конечностей, дизартрией и интеллектуальным снижением [Hughes С. et al., 2001].
Таким образом, анализ клинико-генетических корреляций в семьях с рецессивными и (в меньшей степени) доминантными спастическими параплегиями показал определенную условность общепринятого подразделения данных заболеваний на «чистые» и осложненные формы [Figlewicz D., Bird Т., 1999; Reid E., 1999]. Более правильным было бы говорить о достаточно обширном спектре спастических синдромов, клиническая картина которых может дивергировать или, напротив, перекрываться как для аллельных, так и для разнолокусных заболеваний.