Хорея Гентингтона. Причины и течение хореи Гентингтона
Гиперкинетическая форма является наиболее типичной и характеризуется началом болезни на 4-6 десятилетии жизни; у больных нарастают размашистые генерализованные хореические гиперкинезы и деменция подкоркового типа, развивающиеся на фоне выраженных эмоционально-волевых нарушений.
Хорея Гентингтона характеризуется неуклонно прогредиептным течением. При ювенильной акинетико-ригидпой форме длительность болезни значительно меньше.
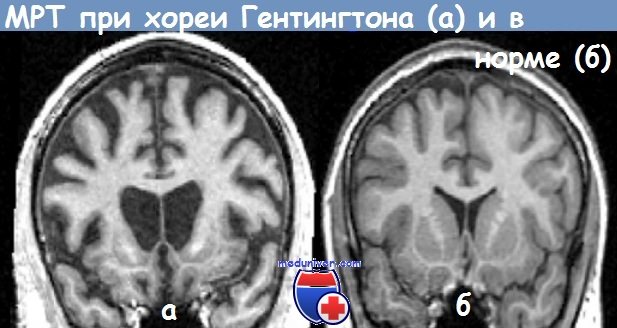
Принимая во внимание выраженный фенотипический полиморфизм хореи Гентингтона, а также неспецифический характер изменений, выявляемых при использовании рутинных лабораторно-инструментальных методов исследования (расширение боковых желудочков и субарахпоидального пространства больших полушарий при КТ и МРТ, депрессия а-ритма на электроэнцефалограмме), важной задачей является внедрение в практику методов ДНК-диагностики данного заболевания.
В 1983 году ген хореи Гентингтона был картирован на коротком плече 4-й хромосомы (локус 4р16.3) в одном из самых первых успешных исследований генетического сцепления у человека [Gusella J. et al., 1983]. Это послужило основой для разработки косвенной пресимптоматической и пренаталыюй ДНК-диагностики хореи Гентингтоыа в информативных семьях [Евграфов О.В. и др., 1991; Иванова-Смоленская И.А. и др., 1992; Meissen G. et al., 1988].
Благодаря накопленному в мире многолетнему опыту такой диагностики и некоторым клинико-генетическим особенностям хореи Гентиигтона (позднее начало, практически полная пенстрантиость мутантного гена, фатальное течение и др.) данное заболевание стало классической «моделью» для изучения различных аспектов медико-генетического консультирования, основанного на ДНК-тестировании.
Предполагается, что в результате происходит формирование патологических полиглутамин-опосредованных связей мутантного гентингтина с молекулами ряда белков ЦНС, что обусловливает его цитотоксичность и в конечном счете селективную гибель нейронов, характерную для хореи Гентингтона [Albin R., Tagle D., 1995; Ross С, 1995]. Такой патогенез болезни подтверждается выявлением в мозге больных хореей Гентингтона характерных внутриядерных включений, содержащих мутантный гентингтин и представляющих собой высокомолекулярные амилоидоподобные агрегаты [Li H. et al., 1995].
Интересно отметить, что лица, имеющие «двойную дозу» мутантного гена (гомозиготные носители патологически удлиненного CAG-участка), клинически неотличимы от больных-гетерозигот по экспансии повторов [Durr A. et al., 1999].
Анализ гена гентингтипа у представителей основных этнических групп мира показал, что 0,75% всех хромосом характеризуются числом CAG-повторов, «промежуточным» между нормой и заведомо патологическими значениями [Kremer В. et al., 1994]. Такие аллели, по-видимому, представляют собой премутацию и служат (в случае дальнейшего нарастания числа повторов) постоянным источником полных мутаций de novo в популяции [Myers R. et al., 1993; Nance M., 1996].
Важно подчеркнуть, что аллели с числом CAG-повторов 36-39 являются «зоной неполной пенетрантности»: часть лиц с указанным числом повторов могут иметь типичные проявления хореи Генгтингтона, тогда как у других носителей таких аллелей заболевание не проявляется даже в самом преклонном возрасте - вплоть до 95 лет [Rubinsztein D. et al., 1996; Brinkman R. et al., 1997; McNeil S. et al., 1997]. Очевидно, что возможность развития заболевания при наличии хромосомы с числом CAG-повторов 36-39 определяется взаимодействием разнообразных экзо- и эндогенных факторов, которые могут иметь как протективное, так и патологическое значение [Nance M., 1996].
В случае наследования хромосом с числом CAG-повторов >40 пенетрантность гена становится полной, и болезнь неизбежно проявится при условии достижения соответствующего возраста.