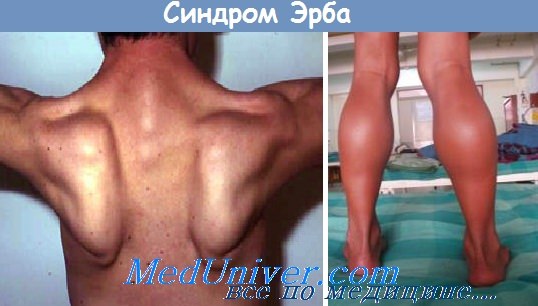
Прогрессирующая мышечная дистрофия Эрба—Рота. Мышечная дистрофия с контрактурами Дрейфуса
Первые сообщения об этом заболевании принадлежат W. Erb (1882) и В. К. Роту (1890). Заболевание дебютирует не только в дошкольном или юношеском возрасте, как предполагали описавшие его исследователи, возможно начало его и в раннем детском возрасте. Наследуется заболевание аутосомно-рецессивно. Первыми симптомами являются слабость и атрофия мышц тазового пояса и проксимальных групп мышц ног. Появляются трудности при подъеме на лестницу, при вставании из положения сидя.
При попытке подняться из положения лежа больной совершает это действие в несколько этапов с помощью рук (вставание «лесенкой»). Позже вовлекаются в патологический процесс мышцы туловища и верхних конечностей. Лопатки выступают, особенно при отведении рук в стороны («крыловидные» лопатки).
Походка больных становится переваливающейся (утиная походка), выражен поясничный лордоз, грудь и живот выпячиваются вперед. Лицо гипомимично («лицо сфинкса») с выступающими губами (губы тапира). Характерна осиная талия. Значительно реже у детей раннего возраста наблюдается нисходящий тип поражения. Динамика распространения патологического процесса у детей раннего возраста лишь начинает выявляться. Более четко она прослеживается в последующие годы.
Болезнь медленно прогрессирует. Однако у детей раннего возраста эта стадия заболевания, как правило, не встречается.
Прогрессирующая мышечная дистрофия с контрактурами Дрейфуса описана J. Drefus в 1928 г. Заболевание наследуется рецессивно, сцеплено с Х-хромосомой, болеют только мальчики. Необычно интенсивное разрастание соединительной ткани в мышцах — отличительная патоморфологическая особенность этой формы патологии.
Заболевание начинается на 3—4-м году жизни с нарастающей слабости в мышцах тазового пояса, позднее присоединяется поражение мышц плечевого пояса. Характерные клинические черты — относительно медленное прогрессирование мышечной слабости и быстрое формирование контрактур. Первыми формируются сгибательные контрактуры локтевых суставов. Укорочение мышц голени и стопы приводит к изменению походки с опорой на большие пальцы. Дистрофические изменения захватывают и сердечную мышцу.
Течение заболевания — медленно прогрессирующее. Эту форму ПМД необходимо дифференцировать с полиартритами, миозитами, деформирующими артрозами.
Под миотоническим феноменом, на основе которого объединяется группа различных по своему геиезу нозологических форм, подразумевается неспособность мышцы быстро расслабляться после однократного сокращения или серии резких сокращений.
На ЭМГ при миотониях находят длительный потенциал последействия, который остается выраженным как при непосредственном раздражении мышцы, так и при воздействии на мышцу через нерв.
Заболевания этой группы включают несколько генетически различных форм: собственно миотонии, миотоническую дистрофию и некоторые другие нозологические формы.
Первичные биохимические дефекты при миотонических синдромах и их патогенез не установлены. У ряда больных обнаружено снижение содержания в мышцах арахидоновой, олеиновой и линолиновой кислот, тогда как содержание жирных кислот С 20 : 2 и С 20 : 3 резко увеличено, что свидетельствует о патологии мембран. В некоторых случаях в крови больных увеличена концентрация калия, может быть слегка повышена утилизация кальция и замедлено его пассивное выведение с мочой. Однако прямой зависимости между этим изменениями и выраженностью миотонического феномена обычно не отмечают.
Характерными для миотонического феномена считаются патоморфологические изменения, обнаруживаемые в терминальной иннервации мышечных волокон. Это могут быть чрезмерные ветвления концевых нервных окончаний или увеличение размеров концевых пластинок. Соответственно концевой иннервации изменяются элементы субневрального аппарата.
Из заболеваний, характеризующихся миотоническим феноменом, у детей раннего возраста встречаются аутосомно-доминантно наследуемая врожденная миотония Томсена, аутосомно-рецессивная миотония и миотоническая дистрофия.