Определение. Заболевание, характеризующееся сочетанием лентигиноза с полипозом ЖКТ, алопецией и дистрофией ногтей.
Историческая справка. В 1955 г. синдром впервые описали американские врачи Leonard W. Cronkhite и Wilma J. Canada. Они выявили у двух женщин сочетание симптомов: универсальное поражение полипами ЖКТ, кожную пигментацию в виде лентиго, алопецию и ониходистрофию.
Этиология и патогенез синдрома Кронкхайта-Канада неизвестны. У пациентов с синдромом Кронкхайта-Канада мутаций гена PTEN не обнаружено. Исследование проводилось в связи с наличием мутаций этого гена у больных с другими синдромами, сопровождающимися полипозом ЖКТ (болезнь Кауден, синдром Рувалкаба—Мюре—Смита и другие). Не исключается в развитии болезни аутоиммунный компонент, что подтверждается выявлением антинуклеарных антител в сыворотке у пациентов с синдромом Кронкхайта-Канада и эффективностью кортикостероидной терапии.
В патогенезе болезни большое значение придается нарушениям пищеварительной, двигательной, всасывающей и секреторной функций кишечника. Это обусловлено полипозом и/или гастроэнтероколитом с образованием воспалительных псевдополипов. Развивающаяся хроническая диарея, способствующая гипопротеинемии, гипокалиемии, потере микроэлементов и аминокислот, по-видимому, является причиной дерматологической симптоматики. Гипопротеинемия (гипоальбуминемия) также возникает при поражении желудка.
Белок теряется вследствие повышенной продукции желудочной слизи и множественных поверхностных некрозов полипов. Данных о семейном характере заболевания нет. Определенную роль в развитии синдрома могут играть тучные клетки, что подтверждается эффективностью терапии стабилизаторами мембраны тучных клеток.
Частота синдрома Кронкхайта-Канада. С 1955 г. по 2002 г. в мировой литературе описано 467 случаев данного синдрома, из них 354 — в Японии. Заболеваемость составляет 1:1 000 000.
Возраст и пол. Клинические проявления отмечаются в диапазоне 31 —86 лет (средний возраст — 55 лет), чаще у мужчин, по одним данным соотношение полов — 1,3—1,5:1, по другим — 3:2.
Изменения кожи, слизистых и придатков кожи проявляются, как правило, через несколько недель или месяцев после возникновения желудочно-кишечных симптомов. Дистрофия ногтей (истончение, размягчение, онихолизис, иногда анонихия) развивается на всех или почти всех пальцах и нередко предшествует желудочно-кишечным симптомам. Пигментация кожи является менее постоянным признаком болезни. Гиперпигментированные пятна имеют размеры от нескольких миллиметров до 10 см.
Обычно они локализуются на коже кистей (крупные пятна — на ладонях, мелкие очаги — на тыльной поверхности), но могут определяться также на лице, шее, нижних конечностях, исключительно редко — на слизистых. На слизистой рта отмечают атрофический глоссит, ангулярный хейлит. Могут регистрироваться алопеция и витилиго.
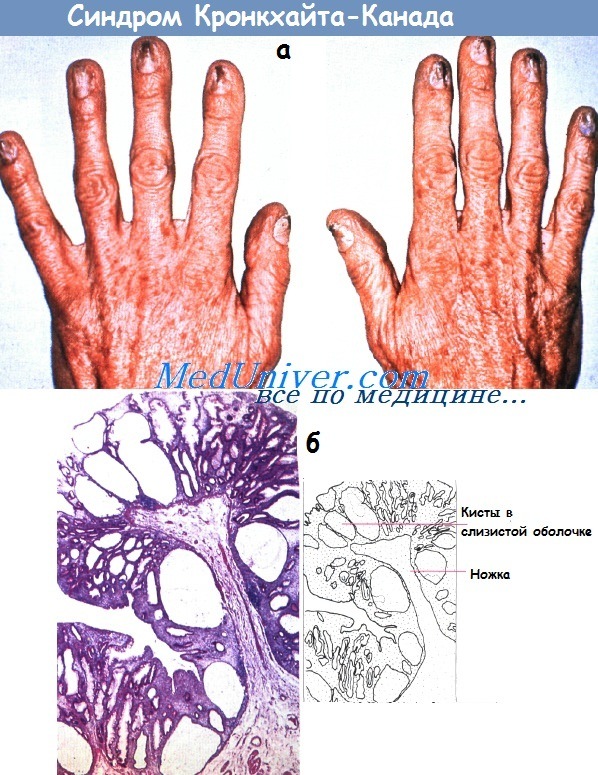
а - Ониходистрофия при синдроме Кронкхайта-Канада.
б - Гистологические проявления синдрома Кронкхайта-Канада.
Кистозные железы не диспластичны и не имеют стромальных элементов, позволяющих отличить их от других форм. Окраска гематоксилин-эозином (х 8).
Изменения ЖКТ. Первые клинические признаки начинаются с постоянных или периодически возникающих болей в области живота, которые сопровождаются водянистыми поносами (5-7 раз в день) и затем через некоторое время кровотечениями. Это приводит к развитию кахексии и отеков. Пациенты за период болезни теряют более 20 кг массы тела. Аденоматозные полипы выявляют на протяжении всего ЖКТ.
Лабораторные показатели. У некоторых больных выявлялись симптомы гипотиреоидизма; в крови — снижение уровня калия, кальция и магния, гипопротеинемия.
Гистология гиперпигментаций кожи. Увеличение количества меланоцитов в базальном слое, очаговый экзоцитоз лимфоцитов в эпидермисе, периваскулярные лимфогистиоцитарные инфильтраты в дерме.
Дифференцируют с заболеваниями, характеризующимися пигментацией кожи, алопецией и дистрофией ногтей.
Прогноз неблагоприятный из-за нередко наблюдаемых желудочно-кишечных кровотечений и возможной трансформации полипов в аденокарциному. Обычно пациенты умирают от желудочно-кишечных кровотечений, сепсиса и сердечной недостаточности; 5-летняя выживаемость составляет 55%. У пациентов с этим синдромом в 13% случаев выявляются злокачественные опухоли ЖКТ: аденокарцинома толстой кишки в 8%, желудка в 5%.
Лечение. Регуляция белкового и минерального обмена веществ с помощью энтеральных добавок, парентерального питания, витаминов, минералов и других средств, а также кортикостероидной терапии. Это помогает в разрешении дерматологических симптомов синдрома: алопеции, гиперпигментации, ониходистрофии и полипоза ЖКТ. Количество полипов уменьшает назначение блокаторов гистаминовых Н2-рецепторов (ранитидин). Выявлена эффективность комбинированной терапии с использованием кромоглигата (стабилизатор мембраны тучных клеток), блокаторов H1- и Н2-рецепторов, кортикостероидов, антибиотиков.
Обнаруживалась эффективность терапии, направленной на лечение Helicobacter pylori: кларитромицин, амоксициллин и омепразол. Хирургическое лечение полипоза. Резекцию сегментов кишечника лучше избегать, ее следует использовать только в случае тяжелых осложнений со стороны ЖКТ.